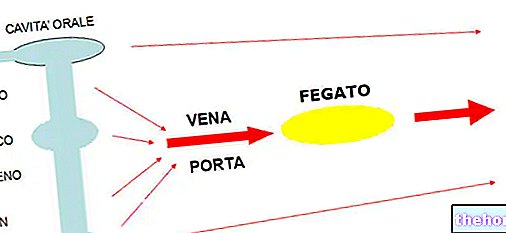
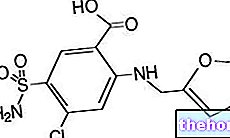
Wirkstoffe: Abirateron (Abirateronacetat)
ZYTIGA 250 mg Tabletten
Warum wird Zytiga verwendet? Wofür ist das?
ZYTIGA enthält ein Arzneimittel namens Abirateronacetat. Es wird verwendet, um Prostatakrebs bei erwachsenen Männern zu behandeln, der sich auf andere Teile des Körpers ausgebreitet hat. ZYTIGA verhindert, dass der Körper Testosteron produziert, was das Wachstum von Prostatakrebs verlangsamen kann.
Wenn Sie dieses Arzneimittel einnehmen, wird Ihr Arzt auch ein anderes Arzneimittel namens Prednison oder Prednisolon verschreiben. Dieses Arzneimittel wird angewendet, um die Möglichkeit von Bluthochdruck, zu viel Wasser im Körper (Flüssigkeitsretention) oder niedrigen Blutspiegeln einer Chemikalie namens Kalium zu verringern.
Kontraindikationen Wann Zytiga nicht angewendet werden sollte
ZYTIGA® darf nicht eingenommen werden
- wenn Sie allergisch gegen Abirateronacetat oder einen der in Abschnitt 6. genannten sonstigen Bestandteile dieses Arzneimittels sind.
- wenn Sie eine Frau sind, insbesondere wenn Sie schwanger sind. Die Anwendung von ZYTIGA ist nur für Männer indiziert.
- wenn Sie eine schwere Leberschädigung haben.
Nehmen Sie dieses Arzneimittel nicht ein, wenn einer dieser Punkte auf Sie zutrifft. Wenn Sie sich nicht sicher sind, fragen Sie vor der Einnahme dieses Arzneimittels Ihren Arzt oder Apotheker.
Vorsichtsmaßnahmen für die Anwendung Was sollten Sie vor der Einnahme von Zytiga® beachten?
Bitte sprechen Sie mit Ihrem Arzt oder Apotheker, bevor Sie dieses Arzneimittel einnehmen:
- wenn Sie Leberprobleme haben
- wenn Ihnen mitgeteilt wurde, dass Sie hohen Blutdruck oder eine Herzinsuffizienz oder einen niedrigen Kaliumspiegel im Blut haben (ein niedriger Kaliumspiegel kann das Risiko für Herzrhythmusstörungen erhöhen).
- wenn Sie jemals andere Probleme mit Ihrem Herzen oder Ihren Blutgefäßen hatten
- wenn Sie einen unregelmäßigen oder schnellen Herzschlag haben
- wenn du kurzatmig bist
- wenn Sie schnell an Gewicht zugenommen haben
- wenn Ihre Füße, Knöchel oder Beine angeschwollen sind
- wenn Sie in der Vergangenheit ein Arzneimittel namens Ketoconazol gegen Prostatakrebs eingenommen haben
- über die Notwendigkeit, dieses Medikament zusammen mit Prednison oder Prednisolon einzunehmen
- über die möglichen Auswirkungen auf die Knochen
- wenn Sie hohe Blutzuckerwerte haben.
Informieren Sie Ihren Arzt, wenn Ihnen bekannt ist, dass Sie Probleme mit Ihrem Herzen oder Ihren Blutgefäßen haben, einschließlich Herzrhythmusstörungen (Arrhythmie) oder wenn Sie mit Arzneimitteln gegen diese Erkrankungen behandelt werden.
Informieren Sie Ihren Arzt, wenn Sie gelbe Haut oder Augen, dunklen Urin oder starke Übelkeit oder Erbrechen haben, da dies Anzeichen oder Symptome von Leberproblemen sein können. In seltenen Fällen kann eine Leberfunktionsstörung (sogenanntes akutes Leberversagen) auftreten, die zum Tod führen kann.
Verminderte rote Blutkörperchen, verminderter Sexualtrieb (Libido), Muskelschwäche und/oder Muskelschmerzen können auftreten.
Wenn Sie sich nicht sicher sind, ob einer der oben genannten Punkte auf Sie zutrifft, sprechen Sie mit Ihrem Arzt oder Apotheker, bevor Sie dieses Arzneimittel einnehmen.
Blutüberwachung
ZYTIGA kann die Leber beeinträchtigen und hat möglicherweise keine Symptome. Wenn Sie dieses Arzneimittel einnehmen, wird Ihr Arzt regelmäßig Blutuntersuchungen durchführen, um zu prüfen, ob sich ZYTIGA auf die Leber auswirkt.
Kinder und Jugendliche
Das Medikament ist bei Kindern und Jugendlichen nicht angezeigt. Wenn ZYTIGA versehentlich von einem Kind oder Jugendlichen eingenommen wird, gehen Sie sofort ins Krankenhaus und nehmen Sie die Packungsbeilage mit, um sie dem Notarzt vorzuzeigen.
Wechselwirkungen Welche Medikamente oder Lebensmittel können die Wirkung von Zytiga® verändern?
Fragen Sie Ihren Arzt oder Apotheker, bevor Sie jedes Arzneimittel einnehmen.
Informieren Sie Ihren Arzt oder Apotheker, wenn Sie andere Arzneimittel einnehmen, kürzlich andere Arzneimittel eingenommen haben oder beabsichtigen andere Arzneimittel einzunehmen. Dies ist wichtig, da ZYTIGA die Wirkung einiger Arzneimittel verstärken kann, einschließlich Herzarzneimitteln, Beruhigungsmitteln, pflanzlichen Arzneimitteln (z. B. Johanniskraut) usw. Ihr Arzt wird möglicherweise entscheiden, die Dosis dieser Arzneimittel zu ändern. Einige Arzneimittel können auch erhöhen oder verringern die Wirkungen von ZYTIGA, die zu Nebenwirkungen führen können oder ZYTIGA möglicherweise nicht so gut wirkt, wie es sollte.
Andere Arzneimittel, die zusammen mit ZYTIGA® eingenommen werden
Eine Androgenentzugsbehandlung kann das Risiko von Herzrhythmusstörungen erhöhen.
Informieren Sie Ihren Arzt, wenn Sie Arzneimittel einnehmen:
- zur Behandlung von Herzrhythmusstörungen (zB Chinidin, Procainamid, Amiodaron und Sotalol);
- bekanntermaßen das Risiko von Herzrhythmusstörungen erhöht [z. B. Methadon (zur Linderung von Schmerzen und zur Behandlung von Drogenabhängigkeit), Moxifloxacin (ein Antibiotikum), Antipsychotika (zur Behandlung schwerer psychischer Erkrankungen)].
ZYTIGA mit Essen
- Dieses Arzneimittel darf nicht zusammen mit Nahrungsmitteln eingenommen werden (siehe Abschnitt „Bei Einnahme dieses Arzneimittels“).
- Die Einnahme von ZYTIGA zusammen mit Nahrungsmitteln kann Nebenwirkungen haben.
Warnungen Es ist wichtig zu wissen, dass:
Schwangerschaft und Stillzeit
Die Anwendung von ZYTIGA ist bei Frauen nicht angezeigt.
- Dieses Arzneimittel kann den Fötus schädigen, wenn es von schwangeren Frauen eingenommen wird.
- Schwangere oder möglicherweise schwangere Frauen sollten Handschuhe tragen, wenn sie ZYTIGA berühren oder handhaben müssen.
- Wenn Sie Sex mit einer Frau im gebärfähigen Alter haben, sollten Sie ein Kondom und eine andere "wirksame Verhütungsmaßnahme" verwenden. Wenn Sie Sex mit einer schwangeren Frau haben, verwenden Sie zum Schutz des Fötus ein Kondom.
Verkehrstüchtigkeit und das Bedienen von Maschinen
Es ist unwahrscheinlich, dass dieses Arzneimittel Ihre Verkehrstüchtigkeit und Ihre Fähigkeit zum Bedienen von Werkzeugen oder Maschinen beeinträchtigt.
ZYTIGA enthält Lactose und Natrium
- ZYTIGA enthält Lactose (eine Zuckerart). Bitte nehmen Sie dieses Arzneimittel erst nach Rücksprache mit Ihrem Arzt ein, wenn Ihnen bekannt ist, dass Sie unter einer Zuckerunverträglichkeit leiden.
- Dieses Arzneimittel enthält ungefähr 27 mg Natrium in einer Tagesdosis von vier Tabletten. Dies sollte bei Patienten mit einer natriumreduzierten Diät berücksichtigt werden.
Dosis, Methode und Zeitpunkt der Verabreichung Wie ist Zytiga anzuwenden: Dosierung
Nehmen Sie dieses Arzneimittel immer genau nach Absprache mit Ihrem Arzt ein. Wenn Sie sich nicht sicher sind, wenden Sie sich an Ihren Arzt oder Apotheker.
Wie viel nehmen
Die empfohlene Dosis beträgt 1.000 mg (vier Tabletten) einmal täglich.
Einnahme dieses Arzneimittels
- Nehmen Sie dieses Arzneimittel oral ein.
- Nehmen Sie ZYTIGA nicht mit Nahrung ein.
- Nehmen Sie ZYTIGA mindestens zwei Stunden nach einer Mahlzeit ein und essen Sie nach der Einnahme von ZYTIGA mindestens eine Stunde lang nichts (siehe Abschnitt 2 „ZYTIGA zusammen mit einer Mahlzeit“).
- Schlucken Sie die Tabletten im Ganzen mit etwas Wasser.
- Brechen Sie die Tabletten nicht.
- ZYTIGA wird zusammen mit einem Arzneimittel namens Prednison oder Prednisolon eingenommen. Nehmen Sie Prednison oder Prednisolon genau nach den Anweisungen Ihres Arztes ein.
- Sie müssen während der Einnahme von ZYTIGA täglich Prednison oder Prednisolon einnehmen.
- Im Notfall muss die Menge von Prednison oder Prednisolon möglicherweise geändert werden. Ihr Arzt wird Sie beraten, wenn Sie die Menge an Prednison oder Prednisolon, die Sie einnehmen, ändern müssen. Brechen Sie die Einnahme von Prednison oder Prednisolon nicht ab, es sei denn, Ihr Arzt sagt es Ihnen.
Ihr Arzt kann Ihnen auch andere Arzneimittel verschreiben, während Sie ZYTIGA und Prednison oder Prednisolon einnehmen.
Wenn Sie die Einnahme von ZYTIGA® vergessen haben
- Wenn Sie die Einnahme von ZYTIGA oder Prednison oder Prednisolon vergessen haben, nehmen Sie am nächsten Tag Ihre übliche Dosis ein.
- Wenn Sie die Einnahme von ZYTIGA oder Prednison oder Prednisolon länger als einen Tag vergessen haben, sprechen Sie mit Ihrem Arzt, ohne zu lange zu warten.
Wenn Sie die Einnahme von ZYTIGA® abbrechen
Brechen Sie die Einnahme von ZYTIGA oder Prednison oder Prednisolon nicht ab, es sei denn, Ihr Arzt sagt es Ihnen.
Wenn Sie weitere Fragen zur Anwendung dieses Arzneimittels haben, wenden Sie sich an Ihren Arzt oder Apotheker.
Überdosierung Was ist zu tun, wenn Sie zu viel Zytiga eingenommen haben?
Wenn Sie eine größere Menge von ZYTIGA eingenommen haben, als Sie sollten, sprechen Sie sofort mit Ihrem Arzt oder suchen Sie sofort ein Krankenhaus auf.
Nebenwirkungen Was sind die Nebenwirkungen von Zytiga
Wie alle Arzneimittel kann auch dieses Arzneimittel Nebenwirkungen haben, die aber nicht bei jedem auftreten müssen.
Wenn Sie eine der in dieser Packungsbeilage aufgeführten Nebenwirkungen bemerken, beenden Sie die Einnahme von ZYTIGA und wenden Sie sich sofort an einen Arzt:
- Muskelschwäche, Muskelkrämpfe oder Herzklopfen (Palpitationen). Dies können Anzeichen für einen niedrigen Kaliumspiegel im Blut sein.
Andere Nebenwirkungen sind:
Sehr häufig (kann mehr als 1 von 10 Patienten betreffen)
Flüssigkeit in den Beinen oder Füßen, niedriger Kaliumspiegel im Blut, hoher Blutdruck, Harnwegsinfektion, Durchfall.
Häufig (kann bis zu 1 von 10 Patienten betreffen)
Hohe Blutfettwerte, erhöhte Leberfunktionswerte, Brustschmerzen, Herzrhythmusstörungen, Herzinsuffizienz, schneller Herzrhythmus, schwere Infektion namens Sepsis, Knochenbrüche, Verdauungsstörungen, Blut im Urin, Hautausschlag.
Gelegentlich (kann bis zu 1 von 100 Patienten betreffen)
Probleme mit Ihren Nebennieren (im Zusammenhang mit Salz- und Wasserproblemen), Muskelschwäche und / oder Muskelschmerzen.
Selten (kann bis zu 1 von 1.000 Patienten betreffen)
Lungenreizung (auch allergische Alveolitis genannt). Probleme mit der Leberfunktion (auch akutes Leberversagen genannt).
Nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar)
Herzinfarkt, Veränderungen im EKG - Elektrokardiogramm (QT-Verlängerung).
Knochenschwund kann bei Männern auftreten, die wegen Prostatakrebs behandelt werden. ZYTIGA in Kombination mit Prednison oder Prednisolon kann den Knochenabbau verstärken.
Meldung von Nebenwirkungen
Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt oder Apotheker, einschließlich aller möglichen Nebenwirkungen, die nicht in dieser Packungsbeilage aufgeführt sind. Sie können Nebenwirkungen auch direkt über das in Anhang V aufgeführte nationale Meldesystem melden. Indem Sie Nebenwirkungen melden, können Sie dazu beitragen, dass mehr Informationen über die Sicherheit dieses Arzneimittels zur Verfügung gestellt werden.
Ablauf und Aufbewahrung
- Bewahren Sie dieses Arzneimittel für Kinder unzugänglich auf.
- Verwenden Sie dieses Arzneimittel nicht nach dem auf dem Karton- und Flaschenetikett angegebenen Verfallsdatum Das Verfallsdatum bezieht sich auf den letzten Tag dieses Monats.
- Unter 30 °C lagern.
- Werfen Sie Arzneimittel nicht ins Wasser oder in den Hausmüll. Fragen Sie Ihren Apotheker, wie Sie Arzneimittel, die Sie nicht mehr verwenden, entsorgen sollen, um die Umwelt zu schützen.
Frist "> Weitere Informationen
Was ZYTIGA enthält
- Der Wirkstoff ist Abirateronacetat. Jede Tablette enthält 250 mg Abirateronacetat.
- Die sonstigen Bestandteile sind: Mikrokristalline Cellulose, Croscarmellose-Natrium, Lactose-Monohydrat; Magnesiumstearat, Povidon (K29 / K32), wasserfreies kolloidales Siliciumdioxid und Natriumlaurylsulfat (siehe Abschnitt 2 „ZYTIGA enthält Lactose und Natrium“).
Beschreibung wie ZYTIGA aussieht und Inhalt der Packung
- ZYTIGA Tabletten sind oval, weiß bis cremefarben, mit der Prägung „AA250“ auf einer Seite.
- Die Tabletten werden in einer Plastikflasche mit einem kindersicheren Plastikverschluss geliefert. Jede Flasche enthält 120 Tabletten. Jede Box enthält eine Flasche.
Quelle Packungsbeilage: AIFA (Italienische Arzneimittelbehörde). Im Januar 2016 veröffentlichter Inhalt. Die vorliegenden Informationen können nicht aktuell sein.
Um Zugriff auf die aktuellste Version zu haben, ist es ratsam, auf die Website der AIFA (Italienische Arzneimittelbehörde) zuzugreifen. Haftungsausschluss und nützliche Informationen.
01.0 BEZEICHNUNG DES ARZNEIMITTELS -
ZYTIGA 250 mg Tabletten
02.0 QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG -
Jede Tablette enthält 250 mg Abirateronacetat.
Hilfsstoffe mit bekannter Wirkung
Jede Tablette enthält 189 mg Lactose und 6,8 mg Natrium.
Die vollständige Auflistung der sonstigen Bestandteile finden Sie in Abschnitt 6.1.
03.0 DARREICHUNGSFORM -
Tablette
Weiße bis cremefarbene, ovale Tabletten (15,9 mm lang x 9,5 mm breit), mit der Prägung AA250 auf einer Seite.
04.0 KLINISCHE INFORMATIONEN -
04.1 Anwendungsgebiete -
ZYTIGA wird zusammen mit Prednison oder Prednisolon angezeigt bei:
• die Behandlung des metastasierten kastrationsresistenten Prostatakarzinoms bei asymptomatischen oder leicht symptomatischen erwachsenen Männern nach Versagen einer Androgenentzugstherapie und für die eine Chemotherapie noch nicht klinisch indiziert ist (siehe Abschnitt 5.1).
• die Behandlung von metastasiertem kastrationsresistentem Prostatakrebs bei erwachsenen Männern, deren Erkrankung während oder nach einer Chemotherapie auf Docetaxel-Basis fortgeschritten ist.
04.2 Dosierung und Art der Anwendung -
Dieses Arzneimittel sollte von einem Arzt verschrieben werden, der Erfahrung in der Anwendung von Krebstherapien hat.
Dosierung
Die empfohlene Dosis beträgt 1.000 mg (vier 250-mg-Tabletten) und wird auf nüchternen Magen als einmalige Tagesdosis eingenommen (siehe „Art der Anwendung“ weiter unten). Die Einnahme der Tabletten zusammen mit Nahrung führt zu einer Erhöhung der systemischen Exposition gegenüber Abirateron (siehe Abschnitte 4.5 und 5.2).
ZYTIGA sollte mit einer niedrigen Dosis Prednison oder Prednisolon eingenommen werden. Die empfohlene Dosis von Prednison oder Prednisolon beträgt 10 mg pro Tag.
Medizinische Kastration mit einem Analogon des Gonadotropin-Releasing-Faktors (luteinisierendes Hormon freisetzendes Hormon, LHRH) sollte während der Behandlung bei nicht chirurgisch kastrierten Patienten fortgesetzt werden.
Vor Beginn der Behandlung sollten die Serumtransaminasespiegel in den ersten drei Behandlungsmonaten alle zwei Wochen und danach monatlich gemessen werden. Überwachen Sie monatlich Blutdruck, Serumkalium und Flüssigkeitsretention (siehe Abschnitt 4.4). Patienten mit einem signifikanten Risiko einer kongestiven Herzinsuffizienz sollten jedoch in den ersten drei Behandlungsmonaten alle 2 Wochen und danach monatlich überwacht werden (siehe Abschnitt 4.4).
Erwägen Sie, bei Patienten mit vorbestehender Hypokaliämie oder bei Patienten, die während der Behandlung mit ZYTIGA eine Hypokaliämie entwickeln, einen Kaliumspiegel von ≥ 4,0 mM aufrechtzuerhalten.
Bei Patienten, die Toxizitäten vom Grad ≥ 3 entwickeln, einschließlich Hypertonie, Hypokaliämie, Ödeme und andere nicht mineralokortikoide Toxizitäten, sollte die Behandlung abgebrochen und eine geeignete Therapie eingeleitet werden. Die Behandlung mit ZYTIGA sollte erst wieder aufgenommen werden, wenn die Toxizitätssymptome auf Grad 1 oder den Ausgangswert zurückgegangen sind.
Wenn eine Tagesdosis von ZYTIGA, Prednison oder Prednisolon vergessen wurde, sollte die Behandlung am nächsten Tag mit der üblichen Tagesdosis wieder aufgenommen werden.
Hepatotoxizität
Bei Patienten, die während der Behandlung eine Hepatotoxizität entwickeln (Anstieg der Alanin-Aminotransferase [ALT] oder Aspartat-Aminotransferase [AST] um mehr als das 5-Fache des oberen Normwertes [ULN]), sollte die Behandlung sofort abgebrochen werden (siehe Abschnitt 4.4). Die Wiederaufnahme der Behandlung, nachdem die Leberfunktionstestwerte des Patienten auf den Ausgangswert zurückgekehrt sind, kann mit einer reduzierten Dosis von 500 mg (zwei Tabletten) einmal täglich erfolgen. Bei Patienten, die sich einer erneuten Behandlung unterziehen, sollten die Serumtransaminasespiegel drei Monate lang mindestens alle zwei Wochen und danach jeden Monat kontrolliert werden. Sollte die Hepatotoxizität mit der reduzierten Dosis von 500 mg pro Tag erneut auftreten, die Behandlung muss abgebrochen werden.
Wenn Patienten zu irgendeinem Zeitpunkt während der Therapie eine schwere Hepatotoxizität entwickeln (ALT oder AST 20 mal ULN), sollte die Behandlung abgebrochen und die Patienten nicht erneut behandelt werden.
Leberfunktionsstörung
Bei Patienten mit vorbestehender leichter Leberfunktionsstörung, Child-Pugh-Klasse A, ist keine Dosisanpassung erforderlich.
Eine mäßige Leberfunktionsstörung (Child-Pugh-Klasse B) führt nach oralen Einzeldosen von 1.000 mg Abirateronacetat zu einer etwa vierfachen Erhöhung der systemischen Exposition gegenüber Abirateron (siehe Abschnitt 5.2). von Abirateronacetat, wenn es Patienten mit mittelschwerer oder schwerer Leberfunktionsstörung (Child-Plugh-Klasse B oder C) verabreicht wird. Eine Dosisanpassung ist nicht zu erwarten. Die Anwendung von ZYTIGA sollte bei Patienten mit mäßiger Leberfunktionsstörung, bei denen der Nutzen das mögliche Risiko deutlich überwiegen muss, mit Vorsicht erwogen werden (siehe Abschnitte 4.2 und 5.2).ZYTIGA sollte bei Patienten mit schwerer Leberfunktionsstörung nicht angewendet werden (siehe Abschnitte 4.3, 4.4 .). und 5.2).
Nierenfunktionsstörung
Bei Patienten mit eingeschränkter Nierenfunktion ist keine Dosisanpassung erforderlich (siehe Abschnitt 5.2). Es liegen jedoch keine klinischen Erfahrungen bei Patienten mit Prostatakrebs und schwerer Nierenfunktionsstörung vor. Bei diesen Patienten ist Vorsicht geboten (siehe Abschnitt 4.4).
Kinder und Jugendliche
Es gibt keine Indikation für eine spezifische Anwendung von ZYTIGA bei Kindern und Jugendlichen.
Art der Verabreichung
ZYTIGA ist zur oralen Anwendung bestimmt.
Die Tabletten sollten mindestens zwei Stunden nach einer Mahlzeit eingenommen werden und mindestens eine Stunde nach Einnahme der Tabletten darf keine Nahrung mehr eingenommen werden.Die Tabletten sollten unzerkaut mit etwas Wasser geschluckt werden.
04.3 Kontraindikationen -
- Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
- Frauen, die schwanger sind oder im gebärfähigen Alter sind (siehe Abschnitt 4.6).
• Schwere Leberfunktionsstörung [Child-Plugh-Klasse-C-Skala (siehe Abschnitte 4.2, 4.4 und 5.2)].
04.4 Besondere Warnhinweise und geeignete Vorsichtsmaßnahmen für die Anwendung -
Hypertonie, Hypokaliämie, Flüssigkeitsretention und Herzinsuffizienz durch einen Überschuss an Mineralokortikoiden
ZYTIGA kann als Folge der durch die CYP17-Hemmung verursachten erhöhten Mineralocorticoidspiegel (siehe Abschnitt 5.1) zu Hypertonie, Hypokaliämie und Flüssigkeitsretention führen (siehe Abschnitt 4.8). Die gleichzeitige Anwendung eines Kortikosteroids hemmt die Aktivität des adrenokortikotropen Hormons (ACTH), was zu einer Verringerung der Häufigkeit und Schwere dieser Nebenwirkungen führt. Vorsicht ist geboten bei der Behandlung von Patienten mit zugrunde liegenden klinischen Zuständen, die durch einen Anstieg des Blutdrucks beeinträchtigt werden können , von Hypokaliämie (z. B. Patienten, die mit Herzglykosiden behandelt werden) oder Flüssigkeitsretention (z. B. Patienten mit Herzinsuffizienz), mit schwerer oder instabiler Angina pectoris, kürzlich aufgetretenem Myokardinfarkt oder ventrikulärer Arrhythmie und Patienten mit eingeschränkter Nierenfunktion.
ZYTIGA sollte bei Patienten mit kardiovaskulären Erkrankungen in der Vorgeschichte mit Vorsicht angewendet werden. Klinische Studien der Phase 3 schlossen Patienten mit unkontrollierter Hypertonie, klinisch signifikanter Herzerkrankung mit Myokardinfarkt oder atherothrombotischen Ereignissen innerhalb der letzten 6 Monate, schwerer oder instabiler Angina pectoris oder Herzinsuffizienz der Klasse III oder IV aus New Yorker Herzverband (NYHA) (Studie 301) oder Herzinsuffizienz der Klasse II - IV (Studie 302) oder Messung der kardialen Ejektionsfraktion Vorhofflimmern oder andere Herzrhythmusstörungen, die eine medikamentöse Therapie erfordern Sicherheit bei Patienten mit linksventrikulärer Ejektionsfraktion (LVEF)
Vor der Behandlung von Patienten mit einem signifikanten Risiko einer kongestiven Herzinsuffizienz (z. B. Herzinsuffizienz in der Anamnese, unkontrollierter Bluthochdruck oder kardiale Ereignisse wie ischämische Herzkrankheit) sollte eine Beurteilung der Herzfunktion (z. B. Echokardiogramm) in Erwägung gezogen werden Herzinsuffizienz und optimierte Herzfunktion Bluthochdruck, Hypokaliämie und Flüssigkeitsretention sollten korrigiert und kontrolliert werden. Während der Behandlung sollten Blutdruck, Serumkalium und Flüssigkeitsretention (Gewichtszunahme, periphere Ödeme) und alle anderen Anzeichen und Symptome einer kongestiven Herzinsuffizienz alle 2 Wochen für 3 Monate und dann monatlich und auf Auffälligkeiten korrigiert. Bei Patienten mit Hypokaliämie wurde im Zusammenhang mit einer Behandlung mit ZYTIGA eine Verlängerung des QT-Intervalls beobachtet. Bewerten Sie die Herzfunktion wie klinisch angezeigt, leiten Sie eine geeignete Behandlung ein und erwägen Sie, diese Behandlung im Falle einer signifikanten Abnahme der Herzfunktion abzubrechen (siehe Abschnitt 4.2).
Hepatotoxizität und Leberfunktionsstörung
In kontrollierten klinischen Studien wurden deutliche Erhöhungen der Leberenzyme beobachtet, die zu einer Unterbrechung der Behandlung oder Dosisanpassungen führten (siehe Abschnitt 4.8).Serumtransaminasespiegel sollten alle zwei Wochen vor Beginn der Behandlung gemessen werden, in den ersten drei Monaten der Behandlung und jeden Monat danach. Wenn klinische Anzeichen und Symptome auftreten, die auf eine Hepatotoxizität hindeuten, sollten die Serumtransaminasen sofort gemessen werden. Sollten ALT oder AST zu irgendeinem Zeitpunkt um das 5-fache l "ULN ansteigen, sollte die Behandlung sofort abgebrochen und die Leberfunktion engmaschig überwacht werden . Die Behandlung kann mit einer reduzierten Dosis erst wieder aufgenommen werden, nachdem die Leberfunktionstestwerte des Patienten auf den Ausgangswert zurückgekehrt sind (siehe Abschnitt 4.2).
Wenn Patienten zu irgendeinem Zeitpunkt während der Therapie eine schwere Hepatotoxizität entwickeln (ALT- oder AST-Anstieg auf das 20-fache des ULN), sollte die Behandlung abgebrochen und diese Patienten nicht erneut behandelt werden.
Patienten mit aktiver oder symptomatischer Virushepatitis wurden von klinischen Studien ausgeschlossen; Daher gibt es keine Daten, die die Anwendung von ZYTIGA bei dieser Population unterstützen.
Es liegen keine Daten zur klinischen Sicherheit und Wirksamkeit von Mehrfachdosen von Abirateronacetat bei Patienten mit mittelschwerer oder schwerer Leberfunktionsstörung (Child-Plugh-Klasse B oder C) vor. Die Anwendung von ZYTIGA ist bei Patienten mit Vorsicht zu prüfen mittelschwere Leberfunktionsstörung, bei der der Nutzen das mögliche Risiko deutlich überwiegen muss (siehe Abschnitte 4.2 und 5.2). ZYTIGA darf bei Patienten mit schwerer Leberfunktionsstörung nicht angewendet werden (siehe Abschnitte 4.2, 4.3 und 5.2).
Nach Markteinführung gab es seltene Berichte über akutes Leberversagen und fulminante Hepatitis, einige davon mit tödlichem Ausgang (siehe Abschnitt 4.8).
Absetzen der Kortikosteroidgabe und Behandlung von Stresssituationen
Vorsicht und Überwachung auf Nebennierenrindeninsuffizienz wird empfohlen, wenn Patienten die Behandlung mit Prednison oder Prednisolon abbrechen.Wenn ZYTIGA nach dem Absetzen von Kortikosteroiden fortgesetzt wird, sollten die Patienten auf Symptome eines Mineralokortikoidenüberschusses überwacht werden (siehe Informationen oben).
Bei Patienten, die Prednison oder Prednisolon einnehmen und ungewöhnlichem Stress ausgesetzt sind, kann eine Erhöhung der Kortikosteroiddosis vor, während und nach der Stresssituation empfohlen werden.
Knochendichte
Bei Männern mit metastasiertem fortgeschrittenem Prostatakrebs (kastrationsresistenter Prostatakrebs) kann eine Abnahme der Knochendichte auftreten. Die Anwendung von ZYTIGA in Kombination mit einem Glukokortikoid kann diese Wirkung verstärken.
Vorherige Verwendung von Ketoconazol
Patienten mit Prostatakrebs, die zuvor mit Ketoconazol behandelt wurden, können niedrigere Ansprechraten erreichen.
Hyperglykämie
Die Anwendung von Glukokortikoiden kann die Hyperglykämie erhöhen, daher sollte der Blutzucker bei Patienten mit Diabetes häufig gemessen werden.
Anwendung in der Chemotherapie
Die Sicherheit und Wirksamkeit von ZYTIGA bei gleichzeitiger Anwendung mit einer zytotoxischen Chemotherapie sind nicht erwiesen (siehe Abschnitt 5.1).
Unverträglichkeit gegenüber Hilfsstoffen
Dieses Arzneimittel enthält Lactose. Patienten mit der seltenen hereditären Galactose-Intoleranz, Lapp-Lactase-Mangel oder Glucose-Galactose-Malabsorption sollten dieses Arzneimittel nicht einnehmen. Darüber hinaus enthält dieses Arzneimittel mehr als 1 mmol (oder 27,2 mg) Natrium in einer Dosis von vier Tabletten. Dies sollte bei Patienten mit einer natriumreduzierten Diät in Betracht gezogen werden.
Mögliche Risiken
Anämie und sexuelle Dysfunktion können bei Männern mit metastasiertem kastrationsresistentem Prostatakrebs auftreten, einschließlich derer, die mit ZYTIGA behandelt werden.
Auswirkungen auf die Skelettmuskulatur
Bei Patienten, die mit ZYTIGA behandelt wurden, wurden Fälle von Myopathie berichtet. Einige Patienten hatten eine Rhabdomyolyse mit Nierenfunktionsstörung. Die meisten Fälle entwickelten sich innerhalb des ersten Behandlungsmonats und klangen nach Absetzen von ZYTIGA ab. Vorsicht ist geboten bei Patienten, die gleichzeitig mit Arzneimitteln behandelt werden, von denen bekannt ist, dass sie mit Myopathie/Rhabdomyolyse in Zusammenhang stehen.
Wechselwirkungen mit anderen Arzneimitteln
Starke Induktoren von CYP3A4 sollten während der Behandlung vermieden werden, es sei denn, es gibt keine therapeutische Alternative, da das Risiko einer verringerten Exposition gegenüber Abirateron besteht (siehe Abschnitt 4.5).
04.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen -
Wirkung von Nahrungsmitteln auf Abirateronacetat
Die Einnahme mit Nahrung erhöht die Resorption von Abirateronacetat signifikant Die Wirksamkeit und Sicherheit bei Einnahme mit Nahrung ist noch nicht erwiesen, daher sollte dieses Arzneimittel nicht mit Nahrung eingenommen werden (siehe Abschnitte 4.2 und 5.2).
Wechselwirkungen mit anderen Arzneimitteln
Mögliche Beeinflussung der Abirateron-Exposition durch andere Arzneimittel
In einer klinischen pharmakokinetischen Wechselwirkungsstudie an gesunden Probanden, die mit einem starken CYP3A4-Induktor, Rifampicin 600 mg täglich über 6 Tage, gefolgt von einer Einzeldosis von 1.000 mg Abirateronacetat, vorbehandelt wurden, war die mittlere Plasma-AUC von Abirateron um 55, % verringert.
Starke Induktoren von CYP3A4 (z. B. Phenytoin, Carbamazepin, Rifampicin, Rifabutin, Rifapentin, Phenobarbital, Johanniskraut [Hypericum perforatum]) sind während der Behandlung zu vermeiden, es sei denn, es gibt keine therapeutische Alternative.
In einer anderen klinischen pharmakokinetischen Wechselwirkungsstudie an gesunden Probanden hatte die gleichzeitige Gabe von Ketoconazol, einem starken CYP3A4-Inhibitor, keine klinisch signifikante Wirkung auf die Pharmakokinetik von Abirateron.
Potenzial zur Beeinflussung der Exposition anderer Arzneimittel
Abirateron ist ein Inhibitor der Leberenzyme CYP2D6 und CYP2C8.
In einer Studie zur Bestimmung der Wirkungen von Abirateronacetat (plus Prednison) mit einer Einzeldosis des CYP2D6-Substrats Dextromethorphan war die systemische Exposition (AUC) von Dextromethorphan etwa 2,9-fach erhöht. wurde um rund 33 % erhöht.
Vorsicht ist geboten bei der Verabreichung von Arzneimitteln, die durch CYP2D6 aktiviert oder metabolisiert werden, insbesondere Arzneimittel mit niedriger therapeutischer Breite. Eine Dosisreduktion von Arzneimitteln mit einem niedrigen therapeutischen Index, die durch CYP2D6 metabolisiert werden, sollte in Betracht gezogen werden. Beispiele für Arzneimittel, die durch CYP2D6 metabolisiert werden, umfassen Metoprolol, Propranolol, Desipramin, Venlafaxin, Haloperidol, Risperidon, Propafenon, Flecanid, Codein, Oxycodon und Tramadol (die letztgenannten drei Arzneimittel benötigen eine CYP2D6-Aktivität zur Bildung ihrer aktiven analgetischen Metaboliten).
In einer klinischen Wechselwirkungsstudie von CYP2C8 bei gesunden Probanden war die AUC von Pioglitazon um 46 % erhöht und die AUC für M-III und M-IV, die aktiven Metaboliten von Pioglitazon, um 10 % verringert, wenn Pioglitazon verabreicht wurde zusammen mit einer Einzeldosis von 1.000 mg Abirateronacetat Obwohl diese Ergebnisse darauf hinweisen, dass bei Kombination von ZYTIGA mit Arzneimitteln, die hauptsächlich durch CYP2C8 eliminiert werden, keine klinisch signifikanten Erhöhungen der Exposition zu erwarten sind, sollten die Patienten sorgfältig auf Anzeichen von Toxizitäten im Zusammenhang mit CYP2C8 . überwacht werden Substrate mit geringer therapeutischer Breite bei gleichzeitiger Anwendung.
In vitro, die Hauptmetaboliten Abirateronsulfat und N-Oxid Abirateronsulfat hemmen nachweislich den Transporter vonAufnahme hepatisches OATP1B1 und infolgedessen kann dies die Konzentrationen der durch OATP1B1 ausgeschiedenen Arzneimittel erhöhen. Es liegen keine klinischen Daten vor, um die Interaktion mit dem Transporter zu bestätigen.
Anwendung mit Arzneimitteln, von denen bekannt ist, dass sie das QT-Intervall verlängern
Da eine Androgenentzugstherapie das QT-Intervall verlängern kann, ist Vorsicht geboten, wenn ZYTIGA zusammen mit Arzneimitteln angewendet wird, von denen bekannt ist, dass sie das QT-Intervall verlängern, oder Arzneimitteln, die Torsade de Pointes induzieren können, wie Antiarrhythmika der Klasse IA (z. B. Chinidin, Disopyramid) oder Klasse III (z. B. Amiodaron, Sotalol, Dofetilid, Ibutilid), Methadon, Moxifloxacin, Antipsychotika usw.
Anwendung mit Spironolacton
Spironolacton bindet den Androgenrezeptor und kann den Prostata-spezifischen Antigen (PSA)-Spiegel erhöhen. Die Anwendung mit ZYTIGA wird nicht empfohlen (siehe Abschnitt 5.1).
04.6 Schwangerschaft und Stillzeit -
Frauen im gebärfähigen Alter
Es liegen keine Daten zur Anwendung von ZYTIGA bei Schwangeren vor und die Anwendung dieses Arzneimittels ist bei Frauen im gebärfähigen Alter kontraindiziert.
Verhütung bei Männern und Frauen
Es ist nicht bekannt, ob Abirateron oder seine Metaboliten in die Samenflüssigkeit ausgeschieden werden. Wenn die Patientin während der Schwangerschaft Geschlechtsverkehr mit einer Frau hat, wird die Verwendung eines Kondoms empfohlen. Wenn die Patientin Geschlechtsverkehr mit einer Frau im gebärfähigen Alter hat, wird die Verwendung eines Kondoms in Verbindung mit einer anderen wirksamen empfängnisverhütenden Maßnahme empfohlen.Tierstudien haben eine Reproduktionstoxizität gezeigt (siehe Abschnitt 5.3).
Schwangerschaft
ZYTIGA ist bei Frauen nicht angezeigt und während der Schwangerschaft oder bei Frauen im gebärfähigen Alter kontraindiziert (siehe Abschnitte 4.3 und 5.3).
Fütterungszeit
Die Anwendung von ZYTIGA ist bei Frauen kontraindiziert.
Fruchtbarkeit
Abirateron beeinflusst die Fertilität bei männlichen und weiblichen Ratten, aber diese Wirkungen sind vollständig reversibel (siehe Abschnitt 5.3).
04.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen -
ZYTIGA hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
04.8 Nebenwirkungen -
Zusammenfassung des Sicherheitsprofils
Die am häufigsten beobachteten Nebenwirkungen sind periphere Ödeme, Hypokaliämie, Hypertonie und Harnwegsinfektionen.
Andere wichtige Nebenwirkungen sind Herzkrankheiten, Hepatotoxizität, Frakturen und allergische Alveolitis.
ZYTIGA kann als pharmakodynamische Folge des Wirkmechanismus Hypertonie, Hypokaliämie und Flüssigkeitsretention verursachen.In klinischen Studien wurden bei mit Abirateronacetat behandelten Patienten häufiger zu erwartende Nebenwirkungen von Mineralokortikoiden beobachtet als bei Patienten, die mit Placebo behandelt wurden: Hypokaliämie % vs. 11 %, Hypertonie 16 % vs. 11 % und Flüssigkeitsretention (peripheres Ödem) 26 % vs. 20 %. Bei Patienten, die mit Abirateronacetat behandelt wurden, wurden bei 4 % bzw. 2 % der Patienten Hypokaliämie vom Grad 3 und 4 und Hypertonie vom Grad 3 und 4 (Common Terminology Criteria for Adverse Events, CTCAE, Version 3.0) beobachtet. Die Reaktionen von Mineralocorticoiden wurden pharmakologisch mit positiven Ergebnissen behandelt. Die gleichzeitige Anwendung von Kortikosteroiden verringert die Häufigkeit und Schwere dieser Nebenwirkungen (siehe Abschnitt 4.4).
Tabelle der Nebenwirkungen
Studien an Patienten mit fortgeschrittenem metastasiertem Prostatakrebs, die ein LHRH-Analogon erhielten oder sich zuvor einer Orchiektomie unterzogen hatten, umfassten die Verabreichung einer ZYTIGA-Dosis von 1.000 mg pro Tag in Kombination mit einer niedrigen Dosis Prednison oder Prednisolon (10 mg pro .). Tag).
Unerwünschte Arzneimittelwirkungen, die während klinischer Studien und Erfahrungen nach der Markteinführung beobachtet wurden, sind nachfolgend nach Häufigkeitskategorien aufgelistet. Häufigkeitskategorien sind wie folgt definiert: sehr häufig (≥ 1/10); häufig (≥ 1/100,
Innerhalb jeder Häufigkeitskategorie werden die Nebenwirkungen nach abnehmendem Schweregrad berichtet Tabelle 1: In klinischen Studien und Studien nach der Markteinführung festgestellte Nebenwirkungen
* Herzinsuffizienz umfasst auch kongestive Herzinsuffizienz, linksventrikuläre Insuffizienz und verminderte Ejektionsfraktion.
** Frakturen umfassen alle Frakturen außer pathologischen Frakturen
a Spontane Berichte aus der Post-Marketing-Erfahrung
Bei mit Abirateronacetat behandelten Patienten traten die folgenden Nebenwirkungen vom Grad 3 (CTCAE Version 3.0) auf: Hypokaliämie 3 %; Harnwegsinfektion, Alanin-Aminotransferase erhöht, Hypertonie, Aspartat-Aminotransferase erhöht, Frakturen 2%; periphere Ödeme, Herzinsuffizienz und Vorhofflimmern jeweils 1%. Hypertriglyzeridämie Grad 3 und Angina pectoris (CTCAE Version 3.0) traten auf in
Beschreibung ausgewählter Nebenwirkungen
Herz-Kreislauf-Reaktionen
Beide klinischen Phase-3-Studien schlossen Patienten mit unkontrolliertem Bluthochdruck, klinisch signifikanter Herzerkrankung, die durch Myokardinfarkt oder atherothrombotischen Ereignissen innerhalb der letzten 6 Monate, schwerer oder instabiler Angina pectoris oder Herzinsuffizienz der NYHA-Klasse III oder IV (Studie 301) oder Herzinsuffizienz nachgewiesen wurde, aus Klasse II - IV (Studie 302) o Messung der kardialen Ejektionsfraktion, Apoplexie und plötzlichem Herztod In klinischen Studien der Phase 3 betrug die Inzidenz von Nebenwirkungen vom vaskulären Typ bei Patienten, die Abirateronacetat einnahmen, im Vergleich zu Patienten, die Placebo einnahmen: Hypertonie 14,5% gegen 10,5%, Vorhofflimmern 3,4% gegen 3,4%, Tachykardie 2,8% gegen 1,7%, Angina pectoris 1,9% gegen 0,9%, Herzinsuffizienz 1,9% gegen 0,6 % und Arrhythmie 1,1 % gegen 0,4%.
Hepatotoxizität
Bei Patienten, die mit Abirateronacetat behandelt wurden, wurde über Hepatotoxizität mit Erhöhung von ALT, AST und Gesamtbilirubin berichtet.In allen klinischen Studien wurden bei ca. 4 % der Patienten, die Abirateronacetat erhielten, in der Regel während der ersten 3 Monate nach Behandlungsbeginn In der klinischen Studie 301 hatten Patienten mit erhöhten Ausgangswerten von ALT oder AST mit höherer Wahrscheinlichkeit erhöhte Leberwerte als Patienten, die mit normalen Werten begannen. Bei erhöhten ALT oder AST > 5 x ULN oder Bilirubinerhöhungen > 3 x ULN wurden beobachtet, Abirateronacetat wurde abgesetzt oder abgesetzt In zwei Fällen gab es deutliche Erhöhungen bei Leberfunktionstests (siehe Abschnitt 4.4) Zwei Patienten mit normaler Leberfunktion zu Studienbeginn hatten Erhöhungen der ALT oder AST von 15 bis 40 x ULN und in Bilirubin von 2 bis 6 x ULN. Leberfunktionstests bei beiden Patienten normalisierten sich und ein Patient wurde erneut behandelt, ohne dass die Werte erneut angestiegen waren. In Studie 302 wurden bei 35 Patienten (6,5%), die mit Abirateronacetat behandelt wurden, Anstiege von ALT oder AST vom Grad 3 oder 4 beobachtet. Aminotransferase-Erhöhungen verschwanden bei allen außer 3 Patienten (2 mit neuen multiplen Lebermetastasen und 1 mit AST-Erhöhung ungefähr 3 Wochen nach der letzten Dosis von Abirateronacetat.) Behandlungsunterbrechungen aufgrund von ALT-Erhöhungen und AST wurden bei 1,7 % und 1,3 % der Patienten berichtet mit Abirateronacetat und 0,2 % bzw. 0 % der mit Placebo behandelten Patienten; es wurden keine Todesfälle aufgrund von hepatotoxischen Ereignissen berichtet.
In klinischen Studien wurde das Risiko einer Hepatotoxizität durch den Ausschluss von Patienten mit Hepatitis zu Studienbeginn oder mit signifikanten Anomalien der Leberfunktionstests verringert x ULN, Lebermetastasen wurden ausgeschlossen.In der klinischen Studie waren 302 Patienten mit Lebermetastasen nicht geeignet und Patienten mit Ausgangswerten von ALT und AST ≥ 2,5 x ULN wurden ausgeschlossen Abnormale Leberfunktionstests, die bei Patienten, die an klinischen Studien teilnahmen, beobachtet wurden, wurden behandelt dynamisch, indem auf die Unterbrechung der Therapie zurückgegriffen und eine Wiederholung der Behandlung erst zugelassen wird, nachdem die Werte in den Leberfunktionstests auf die Ausgangswerte des Patienten zurückgekehrt sind (siehe Abschnitt 4.2). Patienten mit ALT- oder AST-Erhöhungen > 20 x ULN wurden nicht erneut behandelt. Die Sicherheit einer wiederholten Behandlung bei solchen Patienten ist nicht bekannt. Der Mechanismus der ZYTIGA-assoziierten Hepatotoxizität ist unbekannt.
Meldung von vermuteten Nebenwirkungen
Die Meldung von vermuteten Nebenwirkungen, die nach der Zulassung des Arzneimittels aufgetreten sind, ist wichtig, da sie eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels ermöglicht. Angehörige von Gesundheitsberufen werden gebeten, jeden Verdachtsfall einer Nebenwirkung über das nationale Meldesystem zu melden. "Adresse www. agenziafarmaco.gov.it/it/responsabili.
04.9 Überdosierung -
Es liegen nur begrenzte Erfahrungen mit einer Überdosierung von ZYTIGA beim Menschen vor.
Es gibt kein spezifisches Gegenmittel. Im Falle einer Überdosierung sollte die Einnahme abgebrochen und allgemeine unterstützende Maßnahmen ergriffen werden, einschließlich Überwachung auf Arrhythmien, Hypokaliämie und Anzeichen und Symptome einer Flüssigkeitsretention.Außerdem sollte eine Beurteilung der Leberfunktion durchgeführt werden.
05.0 PHARMAKOLOGISCHE EIGENSCHAFTEN -
05.1 "Pharmakodynamische Eigenschaften -
Pharmakotherapeutische Gruppe: endokrine Therapie, andere Hormonantagonisten und verwandte Wirkstoffe.
ATC-Code: L02BX03.
Wirkmechanismus
Abirateronacetat (ZYTIGA) wird umgewandelt in vivo in Abirateron, einem Inhibitor der Androgenbiosynthese. Insbesondere hemmt Abirateron selektiv das Enzym 17α-Hydroxylase / C17,20-Lyase (CYP17).Dieses Enzym wird normalerweise exprimiert und ist für die Biosynthese von Androgenhormonenin Hoden-, Nebennieren- und neoplastischen Prostatageweben erforderlich.CYP17 katalysiert die Umwandlung von Pregnenolon und Progesteron in Testosteronvorläufer, DHEA bzw. Androstendion, durch 17α-Hydroxylierung und Spaltung der C17,20-Bindung.Die Hemmung von CYP17 verursacht auch eine Erhöhung der Mineralocorticoid-Produktion durch die Nebennieren (siehe Abschnitt 4.4).
Androgen-sensitiver Prostatakrebs spricht auf die Behandlung an, indem er den Androgenspiegel senkt. Androgenentzugstherapien, wie die Behandlung mit LHRH-Analoga oder die Orchiektomie, reduzieren die Produktion von Androgenen in den Hoden, ohne die Produktion von Androgenhormonen in den Nebennieren oder im Tumor zu beeinflussen. Die Behandlung mit ZYTIGA reduziert das Serumtestosteron auf nicht nachweisbare Werte (mittels kommerzieller Tests), wenn es mit LHRH-Analoga verabreicht wird (oder nach Orchiektomie).
Pharmakodynamische Wirkungen
ZYTIGA reduziert Serumtestosteron und andere androgene Hormone auf niedrigere Werte als die, die mit der alleinigen Verwendung von LHRH-Analoga oder Orchiektomie erreicht werden.Dieser Effekt ist die Folge der selektiven Hemmung des CYP17-Enzyms, das für die Biosynthese von Androgenen erforderlich ist. PSA wirkt als Biomarker in Patienten mit Prostatakrebs: In einer klinischen Phase-III-Studie, die bei Patienten durchgeführt wurde, die nach vorangegangener Taxan-Chemotherapie Fortschritte machten, zeigten 38 % der mit Abirateronacetat behandelten Patienten eine Reduktion der PSA-Werte um mindestens 50 % gegenüber dem Ausgangswert gegenüber 10 % der Patienten, die Placebo.
Klinische Wirksamkeit und Sicherheit
Die Wirksamkeit wurde in zwei multizentrischen, randomisierten, placebokontrollierten klinischen Phase-3-Studien (Studien 301 und 302) bei Patienten mit metastasiertem kastrationsresistentem Prostatakrebs nachgewiesen erhielten Docetaxel Die Patienten nahmen ein LHRH-Analogon ein oder hatten sich zuvor einer Orchiektomie unterzogen. Im Wirkstoffbehandlungsarm wurde ZYTIGA in einer Dosis von 1.000 mg pro Tag in Kombination mit einer niedrigen Dosis Prednison oder Prednisolon von 5 mg zweimal täglich verabreicht. Patienten in der Kontrollgruppe erhielten Placebo und eine niedrige Dosis Prednison oder Prednisolon von 5 mg zweimal täglich.
Getrennt festgestellte Variationen der Serum-PSA-Konzentration sind nicht immer prädiktiv für den klinischen Nutzen. Daher wurde in beiden klinischen Studien empfohlen, das Behandlungsschema mit den zugewiesenen Studienbehandlungen beizubehalten, bis die unten aufgeführten Abbruchkriterien für jede klinische Studie erfüllt waren.
In beiden Studien war die Anwendung von Spironolacton nicht erlaubt, da es den Androgenrezeptor bindet und den PSA-Spiegel erhöhen kann.
Studie 302 (Chemotherapie-naive Patienten)
In diese Studie wurden Chemotherapie-naive Patienten eingeschlossen, die asymptomatisch oder leicht symptomatisch waren und für die eine Chemotherapie noch nicht klinisch indiziert war. Eine stärkere Schmerzepisode in den letzten 24 Stunden mit einem Score von 0-1 wurde als asymptomatische Sekunde betrachtet Kurze Schmerzinventur – Kurzform (BPI-SF) und ein Score von 2-3 wurde als leicht symptomatisch angesehen.
In Studie 302 (n = 1088) betrug das mediane Alter der eingeschlossenen Patienten 71 Jahre für die mit ZYTIGA plus Prednison oder Prednisolon behandelten Patienten und 70 Jahre für die mit Placebo plus Prednison oder Prednisolon behandelten Patienten Die Gruppe bestand aus 520 Kaukasiern (95,4 %), 15 Schwarzen (2,8 %), 4 Asiaten (0,7 %) und 6 anderen (1,1 %).Eastern Cooperative Onkology Group (ECOG) betrug 0 bei 76 % der Patienten und 1 bei 24 % der Patienten in beiden Armen. 50 % der Patienten hatten nur Knochenmetastasen, weitere 31 % der Patienten hatten Knochen- und Weichteil- oder Lymphknotenmetastasen und 19 % der Patienten hatten nur Weichteil- oder Lymphknotenmetastasen. Patienten mit viszeralen Metastasen wurden ausgeschlossen. Die Endpunkt primäre Wirksamkeitsscores waren das Gesamtüberleben und das radiologische progressionsfreie Überleben (rPFS). Neben der Größe des Endpunkt Der Nutzen wurde auch anhand der Opioid-Einnahmezeit bei Krebsschmerzen, der Zeit bis zum Beginn der zytotoxischen Chemotherapie, der Zeit bis zur Regression des ECOG-Scores von ≥ 1 Punkt und der Zeit bis zur PSA-Progression basierend auf den Kriterien . bewertet Arbeitsgruppe Prostatakrebs-2 (PCWG2). Die Studienbehandlungen wurden bei eindeutiger klinischer Progression abgebrochen, die Behandlungen konnten nach Ermessen des Prüfarztes auch bei bestätigter radiologischer Progression abgebrochen werden.
Das radiologische progressionsfreie Überleben (rPFS) wurde unter Verwendung von Bildgebung sequentiell gemäß den PCWG2-Kriterien (für Knochenläsionen) und den modifizierten Kriterien von Bewertungskriterien für das Ansprechen bei soliden Tumoren (RECIST) (bei Weichteilverletzungen). Die rPFS-Analysen verwendeten eine zentral überprüfte radiologische Beurteilung der Progression.
Bei der „geplanten Analyse von rPFS c“ gab es 401 Ereignisse, 150 (28%) der mit ZYTIGA behandelten Patienten und 251 (46%) der mit Placebo behandelten Patienten hatten radiologische Anzeichen einer Progression oder waren gestorben. Zwischen den Behandlungsgruppen wurde ein signifikanter Unterschied in rPFS beobachtet (siehe Tabelle 2).
NE = Nicht geschätzt
* P-Wert basierend auf Log-Rank-Test, angepasst an ECOG-Stratifizierungsfaktoren (0 oder 1)
** Gefahrenquote
Die Erhebung der Patientendaten wurde jedoch bis zum Datum der zweiten Analyse fortgesetzt ad interim Gesamtüberleben (Gesamtüberleben - Betriebssystem). Die radiologische Untersuchung von rPFS durch den Prüfarzt ist in Tabelle 3 dargestellt.
Sechshundertsieben Patienten hatten eine radiologische Progression oder starben: 271 (50 %) in der Abirateronacetat-Gruppe und 336 (62 %) in der Placebo-Gruppe. Die Behandlung mit Abirateronacetat reduzierte das Risiko einer radiologischen Progression oder des Todes um 47 % im Vergleich zu Placebo (HR = 0,530; 95 %-KI: [0,451; 0,623], p
* P-Wert basierend auf Log-Rank-Test, angepasst an ECOG-Stratifizierungsfaktoren (0 oder 1)
** Gefahrenquote
Nach der Beobachtung von 333 Todesfällen wurde eine geplante Interimsanalyse (IA) für das OS durchgeführt. Basierend auf dem beobachteten signifikanten klinischen Nutzen wurde die Studie eröffnet und den Patienten in der Placebo-Gruppe eine Behandlung mit ZYTIGA angeboten. Die Gesamtüberlebensdauer von ZYTIGA war länger als mit Placebo mit eine 25-prozentige Reduktion des Sterberisikos (HR = 0,752; 95-%-KI: [0,606; 0,934], p = 0,0097), aber das OS war nicht ausgereift und die Ergebnisse ad interim erreichten nicht die Ziel-Stoppgrenzen für statistische Signifikanz (siehe Tabelle 4). Das Überleben ging nach dieser KI weiter.
Die endgültige geplante OS-Analyse wurde nach der Beobachtung von 741 Todesfällen (mediane Nachbeobachtungszeit von 49 Monaten) durchgeführt.65 % der Patienten (354 von 546) behandelten mit ZYTIGA, verglichen mit 71 % (387 von 542) der Patienten mit Placebo behandelt worden waren, gestorben waren.Ein statistisch signifikanter Vorteil beim OS in der ZYTIGA-Gruppe wurde mit einer 19,4%igen Reduktion des Sterberisikos (HR = 0,806; 95%-KI: [0,697; 0,931] , p = 0,0033) und a mediane OS-Verbesserung von 4,4 Monaten (ZYTIGA 34,7 Monate, Placebo 30,3 Monate) (siehe Tabelle 4) Diese Verbesserung wurde nachgewiesen, obwohl 44 % der Patienten unter Placebo ZYTIGA als Folgetherapie erhielten.
NE = Nicht geschätzt
* P-Wert basierend auf Log-Rank-Test, angepasst an ECOG-Stratifizierungsfaktoren (0 oder 1)
** Gefahrenquote
Zusätzlich zu den beobachteten Verbesserungen des Gesamtüberlebens und des rPFS wurde der Nutzen für die Behandlung mit ZYTIGA . nachgewiesen gegen Placebo in allen Endpunkt sekundär wie folgt:
Zeit bis zur PSA-Progression basierend auf den PCWG2-Kriterien: Die mediane Zeit bis zur PSA-Progression betrug 11,1 Monate bei Patienten, die ZYTIGA erhielten, und 5,6 Monate bei Patienten, die Placebo erhielten (HR = 0,488; 95 %-KI: [0,420; 0,568], p
Zeit bis zur Opioid-Einnahme bei Krebsschmerzen: Die mediane Zeit bis zur Opioid-Einnahme bei durch Prostatakrebs verursachten Schmerzen betrug zum Zeitpunkt der endgültigen Analyse 33,4 Monate bei Patienten, die ZYTIGA erhielten, und 23, 4 Monate bei Patienten, die Placebo erhielten (HR = 0,721; 95% CI: [0,614, 0,846], p
Zeit bis zur zytotoxischen Chemotherapie: Die mediane Zeit bis zur zytotoxischen Chemotherapie betrug 25,2 Monate bei Patienten, die ZYTIGA erhielten, und 16,8 Monate bei Patienten, die Placebo erhielten (HR = 0,580; 95 %-KI: [0,487; 0,691], p
Zeit bis zur Verschlechterung des ECOG-Scores ≥ 1 Punkt: Die mediane Zeit bis zur Verschlechterung des ECOG-Scores ≥ 1 Punkt betrug 12,3 Monate bei Patienten, die ZYTIGA erhielten und 10,9 Monate bei Patienten, die Placebo erhielten (HR = 0,821; 95 %-KI: [0,714; 0,943], p = 0,0053).
Die folgenden Endpunkte zeigten einen statistisch signifikanten Vorteil zugunsten der Behandlung mit ZYTIGA:
Objektives Ansprechen: Das objektive Ansprechen wurde als der Prozentsatz der Patienten mit messbarer Erkrankung definiert, die gemäß den RECIST-Kriterien ein vollständiges oder teilweises Ansprechen erreichten (zu Studienbeginn war eine Lymphknotengröße ≥ 2 cm erforderlich, um als Zielläsion angesehen zu werden). Der Prozentsatz der Patienten mit messbarer Erkrankung zu Studienbeginn mit objektivem Ansprechen betrug 36 % in der ZYTIGA-Gruppe und 16 % in der Placebo-Gruppe (p
Schmerzen: Die Behandlung mit ZYTIGA reduzierte das Risiko einer Progression der mittleren Schmerzintensität signifikant um 18 % im Vergleich zur Placebo-Gruppe (p = 0,0490). Die mediane Zeit bis zur Progression betrug 26,7 Monate in der ZYTIGA-Gruppe und 18,4 Monate in der Placebo-Gruppe.
Zeit bis zur Verschlechterung von FACT-P (Gesamtscore): Die Behandlung mit ZYTIGA reduzierte das Risiko einer Verschlechterung von FACT-P (Gesamtscore) um 22 % im Vergleich zu Placebo (p = 0,0028). Die mediane Zeit bis zur Verschlechterung von FACT-P (Gesamtscore) betrug 12,7 Monate in der ZYTIGA-Gruppe und 8,3 Monate in der Placebo-Gruppe.
Studie 301 (Patienten, die zuvor eine Chemotherapie erhalten haben)
In die Studie 301 wurden Patienten aufgenommen, die zuvor Docetaxel erhalten hatten. Bei den Patienten war während der Behandlung mit Docetaxel keine Progression erforderlich, da eine Toxizität dieser Chemotherapie zu einem Abbruch der Chemotherapie geführt haben könnte. Die Patienten setzten die Studienbehandlungen bis zur PSA-Progression (bestätigter Anstieg um 25 % gegenüber dem Ausgangswert des Patienten / niedrigere Werte) fort, zusammen mit der protokolldefinierten radiologischen Progression und der symptomatischen oder klinischen Progression. Patienten mit einer vorherigen Ketoconazol-Behandlung von Prostatakrebs wurden von dieser Studie ausgeschlossen. L"Endpunkt primäre Wirksamkeit war das Gesamtüberleben.
Das durchschnittliche Alter der eingeschlossenen Patienten betrug 69 Jahre (Bereich 39-95). Die Anzahl der mit ZYTIGA behandelten Patienten nach Rassengruppe betrug 737 Kaukasier (93,2 %), 28 Schwarze (3,5 %), 11 Asiaten (1,4 %) und 14 andere (1,8%) 11% der eingeschlossenen Patienten hatten einen Score von Leistungspunktzahl nach der ECOG-Skala von 2; 70 % zeigten röntgenologische Hinweise auf eine Krankheitsprogression mit oder ohne PSA-Progression; 70 % hatten sich zuvor einer zytotoxischen Chemotherapie unterzogen und 30 % hatten zwei. Bei 11% der mit ZYTIGA behandelten Patienten lagen Lebermetastasen vor.
In einer geplanten Analyse, die nach 552 Todesfällen durchgeführt wurde, waren 42 % (333 von 797) der mit ZYTIGA behandelten Patienten gestorben, verglichen mit 55 % (219 von 398) der mit Placebo behandelten Patienten - das mediane Gesamtüberleben der mit ZYTIGA behandelten Patienten (siehe Tabelle 5).
a p-Wert basierend auf einem Log-Rank-Test, angepasst an ECOG-Stratifizierungsfaktoren (0-1 vs. 2), Schmerzscore (abwesend vs. vorhanden), Anzahl vorangegangener Chemotherapien (1 vs. 2) und Art der Krankheitsprogression (nur PSA versus radiologisch).
b Hazard Ratio basierend auf um Stratifizierungsfaktoren bereinigten Risikomodellen. Risikoverhältnis
In allen Stadien der Beurteilung blieb nach den ersten Behandlungsmonaten ein höherer Anteil der mit ZYTIGA behandelten Patienten am Leben als der Anteil der Patienten, die Placebo erhielten.
Überlebens-Subgruppenanalysen zeigten einen signifikanten Überlebensvorteil für die Behandlung mit ZYTIGA.
Neben der beobachteten Verbesserung des Gesamtüberlebens haben alle Endpunkt Sekundäre Studienteilnehmer sprachen sich für ZYTIGA aus und waren nach Adjustierung für mehrere Studien statistisch signifikant, basierend auf den folgenden Punkten:
Mit ZYTIGA behandelte Patienten hatten eine signifikant höhere Gesamt-PSA-Ansprechrate (definiert als eine Reduktion um ≥ 50 % gegenüber dem Ausgangswert) im Vergleich zu Patienten, die Placebo erhielten, 38 % vs. 10 %, p
Die mittlere Zeit bis zur PSA-Progression betrug 10,2 Monate bei mit ZYTIGA behandelten Patienten und 6,6 Monate bei Patienten, die mit Placebo behandelt wurden (HR = 0,580; 95 %-KI: [0,462; 0,728], p
Das mittlere progressionsfreie Überleben, bestimmt durch radiologische Untersuchung, betrug 5,6 Monate für die mit ZYTIGA behandelten Patienten und 3,6 Monate für die mit Placebo behandelten Patienten (HR = 0,673; 95 %-KI: [0,585; 0,776], p
Schmerzen
Der Prozentsatz der Patienten, die über eine Schmerzlinderung berichteten, war in der ZYTIGA-Gruppe statistisch signifikant höher als in der Placebo-Gruppe (44 % vs. 27 %, p = 0,0002). Responder zur Schmerzlinderung wurde definiert als ein Patient, bei dem der schlimmste Schmerzintensitätsscore gemäß BPI SF in den letzten 24 Stunden um mindestens 30 % gegenüber dem Ausgangswert zurückgegangen war, ohne dass der Analgetikaverbrauchsscore in zwei aufeinanderfolgenden Bewertungen angestiegen war Im Abstand von vier Wochen wurden nur Patienten mit einem Score von ≥ 4 und mindestens einem Post-Baseline-Schmerzscore (N = 512) auf Schmerzlinderung analysiert.
Ein geringerer Prozentsatz der mit ZYTIGA behandelten Patienten hatte nach 6 (22 % vs 28 %), 12 (30 % vs 38 %) und 18 Monaten (35 % vs 46 %) eine Schmerzprogression als Patienten, die Placebo erhielten. Die Schmerzprogression wurde definiert als ein Anstieg des BPI SF-Werts für die schlimmste Schmerzintensität um ≥ 30 % gegenüber dem Ausgangswert in den letzten 24 Stunden, wobei bei zwei aufeinanderfolgenden Besuchen keine Abnahme des Scores für die Anwendung von Analgetika beobachtet wurde, oder ein Anstieg des Scores für die Anwendung von Analgetika um ≥ 30 %, der am zwei aufeinanderfolgende Besuche Die Zeit bis zum Fortschreiten der Schmerzen bis zur 25. Perzentile betrug in der ZYTIGA-Gruppe 7,4 Monate, verglichen mit 4,7 Monaten in der Placebo-Gruppe.
Ereignisse, die das Skelettsystem betreffen
Bei einem geringeren Prozentsatz der Patienten in der ZYTIGA-Gruppe traten nach 6 Monaten (18 % vs. 28 %), 12 Monaten (30 % vs. 40 %) und 18 Monaten (35 % vs. 40 %) skelettsystemische Ereignisse auf als bei Patienten in der Placebo-Gruppe. . In der ZYTIGA-Behandlungsgruppe war die Zeit bis zum ersten skelettalen Ereignis im 25. Perzentil doppelt so lang wie in der Kontrollgruppe mit 9,9 Monaten gegenüber 4,9 Monaten. Ein Ereignis des Skelettsystems wurde als pathologische Fraktur, Rückenmarkkompression, palliative Bestrahlung des Knochens oder Operation am Knochen definiert.
Kinder und Jugendliche
Die Europäische Arzneimittel-Agentur hat auf die Verpflichtung zur Vorlage von Studienergebnissen mit ZYTIGA in allen Untergruppen der pädiatrischen Population bei fortgeschrittenem Prostatakrebs verzichtet (siehe Abschnitt 4.2 für Informationen zur pädiatrischen Anwendung).
05.2 "Pharmakokinetische Eigenschaften -
Nach Verabreichung von Abirateronacetat wurde das pharmakokinetische Profil von Abirateron und Abirateronacetat bei gesunden Probanden, bei Patienten mit fortgeschrittenem metastasiertem Prostatakarzinom und bei Patienten ohne Krebs mit Leber- oder Nierenfunktionsstörung untersucht. Abirateronacetat wird schnell umgewandelt in vivo in Abirateron, einem Inhibitor der Androgenbiosynthese (siehe Abschnitt 5.1).
Absorption
Nach oraler Gabe von Abirateronacetat im Nüchternzustand beträgt die Zeit bis zum Erreichen der maximalen Plasmakonzentration von Abirateron etwa 2 Stunden.
Die Einnahme von Abirateronacetat zusammen mit Nahrung führt im Vergleich zur Einnahme im Nüchternzustand zu einer bis zu 10-fachen [AUC] und bis zu 17-fach höheren [Cmax] höheren mittleren systemischen Abirateron-Exposition, bezogen auf das in der Mahlzeit enthaltene Fett Angesichts der normalen Schwankungen in Inhalt und Zusammensetzung der Mahlzeiten kann die Einnahme von ZYTIGA zu den Mahlzeiten zu sehr unterschiedlichen Expositionen führen. Daher sollte ZYTIGA nicht mit Nahrung eingenommen werden. Es sollte mindestens eine Stunde vor oder mindestens zwei Stunden nach einer Mahlzeit eingenommen werden Die Tabletten sollten im Ganzen mit etwas Wasser geschluckt werden (siehe Abschnitt 4.2).
Verteilung
Die Bindung von 14C-markiertem Abirateron an Plasmaproteine beträgt 99,8 %. Das scheinbare Verteilungsvolumen beträgt ca. 5.630 l, was auf eine ausgedehnte Verteilung von Abirateron im peripheren Gewebe hinweist.
Biotransformation
Nach Verabreichung von 14C-radioaktivem Isotopen-markiertem Abirateronacetat in Kapseln wird Abirateronacetat zu Abirateron hydrolysiert, das dann hauptsächlich in der Leber metabolisiert, einschließlich Sulfatierung, Hydroxylierung und Oxidation, erfolgt. Der größte Teil der im Kreislauf vorhandenen Radioaktivität (ca. 92 %) wurde in Form von Metaboliten von Abirateron gefunden. Zwei Hauptmetaboliten der 15 nachweisbaren, Abirateronsulfat und N-Oxid-Abirateronsulfat, machen jeweils etwa 43 % der Gesamtradioaktivität aus.
Beseitigung
Basierend auf Daten von gesunden Probanden beträgt die mittlere Halbwertszeit von Abirateron im Plasma ca. 15 Std. Nach oraler Verabreichung einer Dosis von 1.000 mg radioaktiven Isotopen-markiertem 14C-Abirateronacetat wurden ca. 88 % der radioaktiven Dosis in den Fäzes wiedergefunden und 5 % circanell-Urin. Die Hauptbestandteile im Stuhl sind unverändertes Abirateronacetat und Abirateron (ungefähr 55 % bzw. 22 % der verabreichten Dosis).
Leberfunktionsstörung
Die Pharmakokinetik von Abirateronacetat wurde bei Patienten mit vorbestehender leichter oder mittelschwerer Leberfunktionsstörung (Child-Pugh-Klasse A bzw. B) und bei gesunden Kontrollpersonen untersucht. Die systemische Exposition gegenüber Abirateron nach einer oralen Einzeldosis von 1.000 mg erhöhte sich bei Patienten mit vorbestehender leichter und mittelschwerer Leberfunktionsstörung um etwa 11 % bzw. 260 %. Die mittlere Halbwertszeit von Abirateron war bei Patienten mit leichter Leberfunktionsstörung auf ca. 18 Stunden und bei Patienten mit mittelschwerer Leberfunktionsstörung auf ca. 19 Stunden verlängert.
In einer anderen klinischen Studie wurde die Pharmakokinetik von Abirateron bei Patienten mit vorbestehender schwerer Leberfunktionsstörung (n = 8) (Child-Pugh-Klasse C) und bei 8 gesunden Kontrollpersonen mit normaler Leberfunktion untersucht. Die AUC von Abirateron war bei Patienten mit schwerer Leberfunktionsstörung im Vergleich zu Patienten mit normaler Leberfunktion um ca. 600 % und der freie Anteil des Arzneimittels um 80 % erhöht.
Bei Patienten mit vorbestehender leichter Leberfunktionsstörung ist keine Dosisanpassung erforderlich.
Die Anwendung von Abirateronacetat sollte bei Patienten mit mäßiger Leberfunktionsstörung, bei denen der Nutzen das mögliche Risiko deutlich überwiegen muss, mit Vorsicht erwogen werden (siehe Abschnitte 4.2 und 4.4).Abirateronacetat sollte bei Patienten mit schwerer Leberfunktionsstörung nicht angewendet werden (siehe Abschnitte 4.2). , 4.3 und 4.4).
Bei Patienten, die während der Behandlung eine Hepatotoxizität entwickeln, kann eine Unterbrechung der Behandlung und eine Dosisanpassung erforderlich sein (siehe Abschnitte 4.2 und 4.4)..
Nierenfunktionsstörung
Die Pharmakokinetik von Abirateronacetat wurde bei Patienten mit terminaler Niereninsuffizienz, die sich einem stabilen Hämodialyseschema unterzogen, mit entsprechenden Kontrollpersonen mit normaler Nierenfunktion verglichen. Die systemische Exposition gegenüber Abirateron nach einer oralen Einzeldosis von 1000 mg war bei dialysepflichtigen Patienten mit terminaler Niereninsuffizienz nicht erhöht. Die Anwendung bei Patienten mit Nierenfunktionsstörung, einschließlich schwerer, erfordert keine Dosisreduktion (siehe Abschnitt 4.2 Es liegen jedoch keine klinischen Erfahrungen vor.) bei Patienten mit Prostatakrebs und schwerer Nierenfunktionsstörung. Bei diesen Patienten ist Vorsicht geboten.
05.3 Präklinische Daten zur Sicherheit -
In allen Toxizitätsstudien an Tieren wurde eine signifikante Verringerung der zirkulierenden Testosteronspiegel beobachtet. Als Ergebnis wurden eine Verringerung des Organgewichts sowie morphologische und/oder histopathologische Veränderungen der Fortpflanzungsorgane sowie der Nebennieren-, Hypophysen- und Brustdrüsen festgestellt. Alle Änderungen zeigten eine vollständige oder teilweise Reversibilität. Die Veränderungen der Fortpflanzungsorgane und der auf Androgenhormone empfindlichen Organe sind mit der Pharmakologie von Abirateron vereinbar. Alle medikamentenbedingten Hormonveränderungen kehrten sich nach einer 4-wöchigen Erholungsphase um oder verschwanden.
In Fertilitätsstudien an männlichen und weiblichen Ratten verringerte Abirateronacetat die Fertilität, ein Effekt, der 4 bis 16 Wochen nach Absetzen von Abirateronacetat vollständig reversibel ist.
In einer Entwicklungstoxizitätsstudie an Ratten beeinflusste Abirateronacetat die Schwangerschaft, einschließlich eines verringerten fetalen Gewichts und Überlebens. Auswirkungen auf die äußeren Genitalien wurden beobachtet, obwohl Abirateronacetat nicht teratogen war.
In diesen Studien zur Fertilität und Entwicklungstoxizität an Ratten korrelierten alle Wirkungen mit der pharmakologischen Aktivität von Abirateronacetat.
Abgesehen von den in allen toxikologischen Studien an Tieren festgestellten Variationen in den Fortpflanzungsorganen lassen die präklinischen Daten auf der Grundlage konventioneller Studien mit Sicherheitspharmakologie, Toxizität bei wiederholter Gabe, Genotoxizität und kanzerogenes Potenzial. Abirateronacetat war in einer 6-monatigen Studie an transgenen Mäusen (Tg.rasH2) nicht krebserregend. In einer 24-monatigen Kanzerogenitätsstudie an Ratten erhöhte Abirateronacetat die Inzidenz von interstitiellen Zellneoplasien in den Hoden Dieser Befund hängt vermutlich mit der pharmakologischen Wirkung von Abirateron zusammen und ist rattenspezifisch. Abirateronacetat war bei weiblichen Ratten nicht krebserregend.
Der Wirkstoff Abirateron stellt eine Gefahr für die Gewässer, insbesondere für Fische, dar.
06.0 PHARMAZEUTISCHE INFORMATIONEN -
06.1 Hilfsstoffe -
Mikrokristalline Cellulose
Croscarmellose-Natrium
Lactose-Monohydrat
Magnesiumstearat
Povidon (K29 / K32)
Wasserfreies kolloidales Siliziumdioxid
Natriumlaurylsulfat
06.2 Inkompatibilität "-
Nicht relevant.
06.3 Gültigkeitsdauer "-
2 Jahre.
06.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung -
Für dieses Arzneimittel sind keine besonderen Lagerungsbedingungen erforderlich.
06.5 Art der unmittelbaren Verpackung und Inhalt des Packstücks -
Runde weiße Flaschen aus Polyethylen hoher Dichte mit kindergesichertem Verschluss aus Polypropylen mit 120 Tabletten. Jede Packung enthält eine Flasche.
06.6 Gebrauchs- und Handhabungshinweise -
Aufgrund des Wirkmechanismus kann dieses Arzneimittel den sich entwickelnden Fötus schädigen, daher sollten Frauen, die schwanger sind oder im gebärfähigen Alter, es nicht ohne Schutz, wie z. B. Handschuhe, handhaben.
Nicht verwendete Arzneimittel und Abfälle aus diesem Arzneimittel müssen gemäß den örtlichen Vorschriften entsorgt werden. Dieses Arzneimittel kann ein Risiko für Gewässer darstellen (siehe Abschnitt 5.3).
07.0 INHABER DER "MARKETING GENEHMIGUNG" -
Janssen-Cilag International NV
Turnhoutseweg 30
B-2340 Bier
Belgien
08.0 NUMMER DER VERMARKTUNGSBERECHTIGUNG -
EU / 1/11/714/001
041427016
09.0 DATUM DER ERSTEN GENEHMIGUNG ODER ERNEUERUNG DER GENEHMIGUNG -
Datum der Erstzulassung: 05. September 2011
Letztes Verlängerungsdatum: 26. Mai 2016
10.0 DATUM DER ÜBERARBEITUNG DES TEXTs -
11/2016






-cause-e-rimedi.jpg)