Allgemeinheit
Osteogenesis imperfecta ist eine angeborene genetische Erkrankung, die nicht an das Geschlecht gebunden ist und für eine gewisse Knochenbrüchigkeit und eine ausgeprägte Neigung zu Frakturen verantwortlich ist.
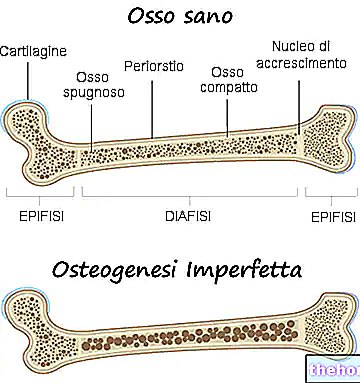
Die Symptome der Osteogenesis imperfecta sind zahlreich; im Allgemeinen bestehen sie aus: Knochenschwächung, hoher Neigung zu Knochenbrüchen, Vorhandensein von blauen, grauen oder violetten Augenskleren, Vorhandensein von Knochendeformitäten oder anderen Skelettveränderungen, dreieckiges Gesicht, Zahnbrüchigkeit usw. .
Im Allgemeinen sind folgende Punkte für eine korrekte Diagnose der Osteogenesis imperfecta unerlässlich: körperliche Untersuchung, Anamnese, medizinische Bildgebungstests, ein Typ-I-Kollagen-Beurteilungstest und ein Gentest.
Leider stehen Patienten mit Osteogenesis imperfecta derzeit nur symptomatische Behandlungen zur Verfügung. Die betreffende Krankheit ist in der Tat unheilbar.
Was ist Osteogenesis imperfecta?
Osteogenesis imperfecta ist eine genetische Erkrankung, die die Knochen des Betroffenen schwächer und anfälliger für Frakturen macht.
Tatsächlich bezeichnen Mediziner mit dem Begriff Osteogenesis imperfecta eine heterogene Gruppe genetischer Erkrankungen, die durch eine gewisse Knochenbrüchigkeit gekennzeichnet sind. Es gibt daher mehrere Formen (oder Typen) der Osteogenesis imperfecta, von denen einige viel schwerwiegender sind als andere.
ES IST EINE ANgeborene KRANKHEIT
Osteogenesis imperfecta ist bei Betroffenen eine von Geburt an bestehende Erkrankung und kann daher im Grunde als angeborene Erkrankung definiert werden.
IST ES SEX-BEZOGEN?
Osteogenesis imperfecta ist keine geschlechtsbedingte genetische Erkrankung wie Hämophilie oder das Klinefelter-Syndrom.
Epidemiologie
Nach einigen statistischen Untersuchungen würde die Inzidenz von Osteogenesis imperfecta einem Fall alle 15.000-20.000 Geburten entsprechen. Dies bedeutet, dass von 15.000-20.000 Neugeborenen eines von Osteogenesis imperfecta betroffen ist.
Andere statistische Studien haben auch gezeigt, dass Osteogenesis imperfecta Männer und Frauen gleichermaßen betrifft und keine Präferenz für eine bestimmte Bevölkerung oder ethnische Gruppe hat.
Die Lebensdauer ist ein äußerst variabler Parameter, der von der Form der Osteogenesis imperfecta abhängt.
Ursachen
Osteogenesis imperfecta resultiert fast immer aus einer qualitativen und quantitativen Veränderung der Produktion von Kollagen Typ I.
Kollagen Typ I ist wichtig für die Stärkung der Knochen und für die Erhaltung eines gesunden Bindegewebes, das Knorpel, Sehnen, Haut, Augensklera usw.
Daher beeinflusst eine Veränderung der Produktion von Kollagen Typ I die Festigkeit der Knochen und die Gesundheit des im menschlichen Körper vorhandenen Bindegewebes.
WAS ÄNDERT DIE KOLLAGENPRODUKTION?
Eine genetische Erkrankung ist ein Zustand, der durch eine Mutation eines oder mehrerer Gene, aus denen die zelluläre DNA besteht, entsteht.
Bei der Osteogenesis imperfecta liegen die Ursachen für letztere fast immer in der Mutation eines oder beider Gene COL1A1 (auf Chromosom 17 lokalisiert) und COL1A2 (auf Chromosom 7 lokalisiert).
Unter normalen Bedingungen regulieren COL1A1 und COL1A2 die normale Produktion von Kollagen Typ I; bei Vorhandensein von Mutationen in ihrer Verantwortung versagen sie in ihrer regulatorischen Funktion.
Wichtig: Welche anderen Gene verursachen, wenn sie mutiert sind, Osteogenesis imperfecta?
Neben den Mutationen von COL1A1 und COL1A2 sind Mutationen in den Genen IFITM5, SERPINF1, CRTAP und LEPRE1 potenzielle Ursachen der Osteogenesis imperfecta.
Die genannten Gene decken andere Funktionen als COL1A1 und COL1A2 ab – kontrollieren also nicht die Produktion von Typ-I-Kollagen – haben aber dennoch einen „Einfluss auf die Festigkeit und Widerstandsfähigkeit der Knochen des menschlichen Skeletts.
WELCHE ART VON GENETISCHEN KRANKHEITEN IST ES?
Osteogenesis imperfecta ist eine autosomal genetisch bedingte Erkrankung.
Der Begriff autosomal, der mit einer genetischen Erkrankung in Verbindung gebracht wird, weist darauf hin, dass die fragliche Erkrankung auf genetische Mutationen zurückzuführen ist, die auf autosomalen und nicht geschlechtsspezifischen Chromosomen basieren.
Die Leser werden daran erinnert, dass der Mensch einen Chromosomensatz von 23 Gesamtchromosomenpaaren besitzt, von denen 22 Paare vom autosomalen Typ und nur ein Paar vom sexuellen Typ sind individuell.
Osteogenesis imperfecta nach Mutationen in COL1A1, COL1A2 und IFITM5 weist alle Merkmale einer autosomal-dominanten Erkrankung auf, wenn sie auf Mutationen in den Genen SERPINF1, CRTAP und LEPRE1 zurückzuführen ist, hat sie die Merkmale einer autosomal-rezessiven Erkrankung.
TYPEN
Derzeit glauben Ärzte, dass es 8 Arten (oder Formen) von Osteogenesis imperfecta gibt. Zur Unterscheidung der verschiedenen Typen entschied man sich für die römische Nummerierung, und zwar für die ersten acht römischen Ziffern.
Die folgende Tabelle zeigt die 8 Formen der Osteogenesis imperfecta, die Mutationen, die sie verursachen und andere Merkmale.
Kerl
Mutiertes Gen
Art der genetischen Erkrankung
DAS
COL1A1
Autosomal dominant
II
COL1A1 und COL1A2
Autosomal dominant
III
COL1A1 und COL1A2
Autosomal dominant
NS
COL1A1 und COL1A2
Autosomal dominant
V.
IFITM5
Autosomal dominant
SIE
SERPINF1
Autosomal-rezessiv
VII
CRTAP
Autosomal-rezessiv
VIII
HASE 1
Autosomal-rezessiv
* Hinweis: Offensichtlich handelt es sich bei den Mutationen in COL1A1 und COL1A2, die die ersten vier Formen der Osteogenesis imperfecta verursachen, um genetische Veränderungen mit leicht unterschiedlichen Merkmalen. Andernfalls würde es keinen Sinn machen, das eine vom anderen zu unterscheiden.
Symptome, Anzeichen und Komplikationen
Alle Arten der Osteogenesis imperfecta sind für eine Schwächung der Knochen verantwortlich, so dass der Betroffene eine besondere Veranlagung für Frakturen hat. Der Schwächungsgrad der Knochen variiert je nach Form; bei einigen von ihnen ist diese Schwächung größer als bei anderen.
Allerdings muss darauf hingewiesen werden, dass jede Form der Osteogenesis imperfecta ihr eigenes symptomatisches Bild hat, das für manche an das symptomatologische Bild anderer Formen erinnern mag.
MÖGLICHE SYMPTOME UND ANZEIGEN
Zu den möglichen Symptomen und Anzeichen einer Osteogenesis imperfecta gehören:
- Vorhandensein von Knochenfehlbildungen;
- Vorhandensein eines kurzen und kleinen Körpers (als Rumpf gedacht);
- Gelenkprobleme (zB: lose Gelenke);
- Muskelschwäche;
- Blaue, violette oder graue Augensklera;
- Dreieckiges Gesicht;
- Fasstruhe;
- Morphologische Anomalien der Wirbelsäule;
- Zerbrechlichkeit der Zähne;
- Hörverlust oder vollständiger Verlust des Hörvermögens;
- Atembeschwerden
- Probleme im Zusammenhang mit dem Fehlen oder Fehlen von Typ-1-Kollagen.
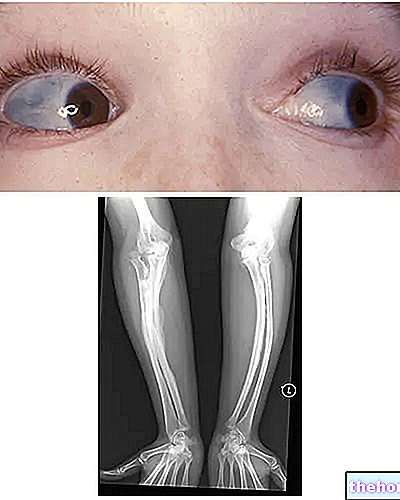
Osteogenesis imperfecta: Beachten Sie die Blaufärbung der Lederhaut und die Knochendeformationen, die die Krankheit charakterisieren. Von wikipedia.org
WELCHES SIND DIE SCHWERSTE FORMEN DER UNVOLLKOMMENEN OSTEOGENESE?
Ärzte klassifizieren den symptomatischen Schweregrad der verschiedenen Arten von Osteogenesis imperfecta auf einer Skala von 3 Grad, nämlich den leichten Grad, den mittleren Grad und den schweren Grad.
Nur eine Form gehört zur Kategorie "leichter Grad": "Typ I Osteogenesis imperfecta"; 4 Formen der Osteogenesis imperfecta gehören zur Kategorie "mittlerer Grad": IV, V und VI; schließlich gehören zur Kategorie "schwerer Grad" 3 Formen: II, III, VII und VIII.
TYP I: MERKMALE
Die häufigste und am wenigsten schwere Form der Osteogenesis imperfecta Typ I weist folgende Merkmale auf:
- Es verursacht vor allem vor der Pubertät Frakturen;
- Es hat "fast keinen Einfluss auf die Körpergröße, daher sind die Patienten normalerweise von normaler" Körpergröße;
- Verursacht Gelenkprobleme und Muskelschwäche
- Es ist für blaue, violette oder graue Lederhaut verantwortlich;
- Es ist die Ursache von dreieckigen Gesichts- und Wirbelsäulenanomalien;
- Es verursacht fast nie Knochendeformitäten. Wenn es sie provoziert, sind sie minimal;
- Es kann Zahnbrüchigkeit und / oder Hörverlust verursachen (letzterer tritt normalerweise im Erwachsenenalter auf);
- Es ist mit dem Vorhandensein von Kollagen Typ I verbunden, das eine normale Qualität, aber eine abnormale Quantität aufweist (es ist schlechter als normal).
TYP II: MERKMALE
Die Osteogenesis imperfecta vom Typ II ist gekennzeichnet durch:
- Todesursache bei der Geburt oder kurz danach. Atemprobleme führen fast immer zum Tod;
- Vorhandensein erheblicher Knochenbrüchigkeit und schwerer Knochendeformitäten;
- Kleinwuchs und unterentwickelte Lungen
- Blaue, violette oder graue Lederhaut;
- Vorhandensein von quantitativen und qualitativen Anomalien von Typ-I-Kollagen.
TYP III: MERKMALE
Die Osteogenesis imperfecta vom Typ III weist folgende Merkmale auf:
- Obwohl es sehr schwerwiegend ist, führt es in der Neugeborenenperiode nicht häufig zum Tod;
- Es wird mit „hoher Knochenbrüchigkeit“ in Verbindung gebracht;
- Es ist verantwortlich für Kleinwuchs, Gelenkprobleme, Muskelschwäche (insbesondere in den Beinen und Armen), Tonnenbrust, dreieckiges Gesicht und abnormale Krümmung der Wirbelsäule;
- Es ist auf blaue, violette oder graue Sklera zurückzuführen;
- Es kann zu Atemproblemen, Zahnbrüchigkeit und Hörverlust führen;
- Es ist häufig für Knochendeformitäten verantwortlich;
- Es ist mit qualitativen und quantitativen Anomalien von Typ-I-Kollagen verbunden.
TYP IV: MERKMALE
Die Osteogenese vom Typ IV ist gekennzeichnet durch:
- Ein Grad an Knochenbrüchigkeit zwischen den Formen II und III und Form I;
- Unterdurchschnittliche Statur;
- Blaue, violette oder graue Lederhaut;
- Knochendeformitäten von leichter / mittelschwerer Entität, leichte Anomalien der Wirbelsäule und des tonnenförmigen Brustkorbs;
- Dreieckiges Gesicht;
- Mögliches Vorhandensein von Zahnbrüchigkeit und Hörverlust;
- Vorhandensein von Typ-I-Kollagenanomalien.
TYP V: EIGENSCHAFTEN
Osteogenesis imperfecta vom Typ V ähnelt in gewisser Weise der Osteogenesis imperfecta vom Typ IV. Es weist jedoch einige Besonderheiten auf, die sind:
- Normal gefärbte Sklera;
- Fehlen von Zahnbrüchigkeit;
- Bildung von abnormen knöchernen Schwielen während des Heilungsprozesses von Knochenbrüchen;
- Verkalkung der Membrana interossea, die sich zwischen Radius und Ulna befindet. Dies beeinträchtigt die Beweglichkeit des Unterarms.
TYP VI: MERKMALE
Auch die Osteogenesis imperfecta vom Typ VI ähnelt der Form IV. Zur Unterscheidung von letzterer sind einige Besonderheiten zu nennen, darunter der hohe Blutspiegel an alkalischer Phosphatase und das Vorhandensein von Lamellen (knöchern) auf einigen Knochen, die den Stacheln von Fischen ähnlich sind.
TYP VII: MERKMALE
Symptomatisch kann die Osteogenesis imperfecta vom Typ VII unter bestimmten Umständen dem Typ IV und unter anderen Umständen dem Typ II ähneln.
Zu den Besonderheiten dieser schwerwiegenden pathologischen Form gehören:
- Kleinwuchs;
- Das Vorhandensein eines extrem kurzen Humerus (Armknochen) und Femur (Oberschenkelknochen);
- Das häufige Auftreten einer Hüftdeformität, bekannt als Coxa vara.
TYP VIII: EIGENSCHAFTEN
Die Osteogenesis imperfecta vom Typ VIII erinnert stark an die Formen II und III.
Unter seinen besonderen Merkmalen sind die folgenden hervorzuheben: das starke Wachstumsdefizit, die schwere Hypomineralisierung des Skeletts und das Fehlen (oder das geringe Vorhandensein) des Prolyl-3-Hydroxylase-Enzyms.
Diagnose
Im Allgemeinen beginnt der diagnostische Prozess, dem Patienten mit Verdacht auf Osteogenesis imperfecta unterzogen werden, mit einer sorgfältigen körperlichen Untersuchung und einer sorgfältigen Anamnese; dann geht es weiter, mit einer "Analyse der Familienanamnese des Patienten und einer Reihe bildgebender Diagnostik (Röntgen, CT-Scans usw.); schließlich endet es mit einer quantitativen und qualitativen Bewertung von Typ-I-Kollagen und mit a genetischer Test.
Heute besteht die Möglichkeit, Osteogenesis imperfecta bereits in der pränatalen Phase zu diagnostizieren, indem man eine Schwangere einem Ultraschall unterzieht.
DIE BEDEUTUNG DER OBJEKTIVEN PRÜFUNG UND DER GESCHICHTE
Ein Facharzt für Osteogenesis imperfecta kann die oben genannte Erkrankung sehr oft auch nur durch eine körperliche Untersuchung und Anamnese diagnostizieren. Dies bedeutet, dass diese diagnostischen Tests von nicht zu vernachlässigender Bedeutung sind.
BEWERTUNG DER PRODUKTION VON TYP I KOLLAGEN
Die qualitative und quantitative Beurteilung von Typ I-Kollagen ist in der Regel ein sehr zuverlässiger Test, da, wie gesagt, die Mehrzahl der Fälle von Osteogenesis imperfecta durch Mutationen in den Genen gekennzeichnet ist, die die Produktion von Typ 1-Kollagen steuern.
Um die Quantität und Qualität von Typ-I-Kollagen auf zellulärer Ebene bei einer Person zu beurteilen, können sich Ärzte auf eine Hautbiopsie oder einen bestimmten Bluttest verlassen.
Beide Bewertungstests sind ziemlich komplex und der Patient (oder seine Eltern) müssen möglicherweise mehrere Wochen warten, um die Ergebnisse zu erfahren.
GENETISCHER TEST
Durch einen genetischen Test, bei dem die gesamte DNA des untersuchten Individuums untersucht wird, können Ärzte die Merkmale der vorliegenden genetischen Mutation endgültig feststellen.
Im Allgemeinen ist die Durchführung eines genetischen Tests an der gesamten zellulären DNA vorgesehen, wenn die Bewertung der Eigenschaften von Kollagen Typ I nicht die gewünschten Ergebnisse erbracht hat oder wenn keine Mutation in COL1A1 oder COL1A2 die " Osteogenesis imperfecta" verursacht.
PRÄNATALDIAGNOSTIK
Pränataler Ultraschall ist sehr nützlich, um Osteogenesis imperfecta vom Typ II und Typ III zu identifizieren.
Therapie
Derzeit gibt es keine spezifische Heilung für Osteogenesis imperfecta, dh Menschen mit Osteogenesis imperfecta sind dazu bestimmt, mit der oben genannten Erkrankung bis zum Tod zu leben, der oft auf die Folgen der Krankheit selbst zurückzuführen ist.
Das Fehlen einer spezifischen Therapie schließt die Existenz anderer Behandlungsformen nicht aus. Tatsächlich gehören zu den therapeutischen Möglichkeiten eines Patienten mit Osteogenesis imperfecta verschiedene symptomatische Therapien; unter symptomatischen Therapien verstehen wir Behandlungen, die in der Lage sind, Symptome zu lindern, den Krankheitsverlauf zu verlangsamen und die schwerwiegendsten Folgen zu verhindern (oder zumindest hinauszuzögern).
MÖGLICHE SYMPTOMATISCHE BEHANDLUNGEN
In der Liste möglicher symptomatischer Behandlungen für Osteogenesis imperfecta ragen folgende heraus:
- Das chirurgische Einsetzen von Nägeln in die längsten Knochen (Anmerkung: am anfälligsten für Frakturen), die eine größere Widerstandsfähigkeit gegen Frakturen und Deformitäten bieten. Diese Operation heißt rudern intramedullär;
- Konservative oder chirurgische Behandlung von Frakturen und / oder Knochendeformitäten;
- Zahnpflege, um die Gesundheit der Zähne zu schützen;
- Schmerzlindernde Therapien bei sehr schmerzhaften Mehrfachfrakturen;
- Physiotherapie zur Muskelverlängerung und -stärkung Ein elastischer und tonisierender Muskelapparat verhindert Stürze, die zu verschiedenen Knochenbrüchen führen können;
- Die Verwendung von Hilfsmitteln zur Fortbewegung wie Rollstühle, Stützen, Krücken etc.
VORTEILE DER BEWEGUNG
Für Menschen mit Osteogenesis imperfecta empfehlen Ärzte im Allgemeinen die ständige Ausübung von körperlicher Bewegung und Bewegung, da beide Aktivitäten zur Stärkung des Skelett- und Muskelsystems beitragen.
Zu den empfohlenen Sportarten gehören: Schwimmen, da es sich um eine "körperliche Aktivität mit geringer Auswirkung auf das Skelettsystem" handelt, und Gehen.
VORTEILE VON EINEM GESUNDEN LEBENSSTIL
Ein gesundes Leben zu führen, das Rauchen zu vermeiden, zu viel Alkohol zu trinken, zu viel und schlecht zu essen usw. hat für Patienten mit Osteogenesis imperfecta mehr als nur einzelne gesundheitliche Vorteile, da es das Fortschreiten der Krankheit verlangsamt und die Knochenbrüchigkeit verringert.
SYMPTOMATISCHE BEHANDLUNGEN IN DER EXPERIMENTATIONSPHASE
Derzeit evaluieren Ärzte und Forscher die Wirksamkeit einiger symptomatischer Behandlungen, einschließlich der Behandlung mit Wachstumshormonen und der intravenösen und oralen Therapie auf Bisphosphonatbasis.
Im Moment sind die Ergebnisse der oben genannten Prüfbehandlungen ein gutes Zeichen für die gesamte medizinische Gemeinschaft.
Prognose
Osteogenesis imperfecta ist eine Krankheit mit negativer Prognose, da sie unheilbar ist, die Lebensqualität drastisch einschränkt und in einigen Fällen zum vorzeitigen Tod des Betroffenen führt.
Es ist jedoch zu beachten, dass auch dank moderner symptomatischer Behandlungen viele Menschen mit einer leichten Form der Osteogenesis imperfecta ein angenehmes und zufriedenes Leben führen können.
Verhütung
Leider gibt es derzeit keine vorbeugende Maßnahme gegen Osteogenesis imperfecta.








.jpg)