Was ist ein Phäochromozytom?
Das Phäochromyzytom ist ein Tumor, der sich in der Nebenniere entwickelt und im Allgemeinen seinen innersten Teil, die sogenannte Medulla, betrifft, in der sich die chromaffinen Zellen befinden. Es handelt sich um eine eher seltene Neubildung mit einer geschätzten Inzidenz zwischen 2 und 8 Fällen pro Million Einwohner, das Phäochromozytom erkennt eine gewisse familiäre Veranlagung an und tritt häufiger bei jungen Erwachsenen und in den mittleren Altersgruppen auf.

Nebennieren und Katecholamine
Die Nebennieren, die wie ein Hut über den Nieren platziert sind, produzieren in ihrem zentralen Teil zwei sehr wichtige Hormone, Adrenalin und Noradrenalin, oder allgemeiner Katecholamine genannt, die zusammen die Reaktion des Körpers auf psychophysischen Stress regulieren und ihn auf den so genannten Stress vorbereiten. Reaktion von Angriff und Flucht genannt. Unter ähnlichen Umständen erhöht das Herz dank der massiven Ausschüttung dieser Hormone die Kraft und die Kontraktionsfrequenz, die Bronchien, die Pupille und die Blutgefäße der Blinddarmmuskeln und des Koronarsystems erweitern sich, während die Glykogenolyse in der Leber stimuliert wird.
Gleichzeitig, immer um den Körper auf die bevorstehende körperliche Aktivität vorzubereiten, werden die Verdauungsprozesse deutlich verlangsamt, während sich die kutanen und peripheren Blutgefäße verengen und der arterielle Druck steigt; die Blase entspannt sich, während sich der Harnröhrenschließmuskel verengt (hemmt das Wasserlassen).
Symptome
Für weitere Informationen: Phäochromozytom-Symptome
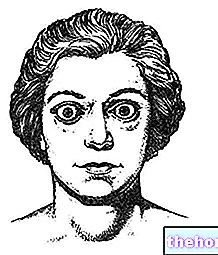
Da es sich beim Phäochromozytom um einen Katecholamin-sezernierenden Tumor handelt, wird er sehr oft von Bluthochdruck (einer grundlegenden klinischen Manifestation) und Symptomen wie plötzlichen Kopfschmerzen, Blässe, starkem Schwitzen, Brustschmerzen, Gewichtsverlust, Nervosität, Angst, Angst, Zittern, Herzklopfen und Gefühl eines energischeren Herzschlags. Manchmal kann es asymptomatisch sein.
Zu den Symptomen eines Phäochromykotoms gehören manchmal auch Magen-Darm-Erkrankungen wie Bauchschmerzen, Übelkeit und Erbrechen sowie ein gestörter Glukosestoffwechsel wie eine gestörte Glukosetoleranz oder ein manifester Diabetes.
In Bezug auf die Katecholaminausschüttung durch den Tumor können die Symptome akzessorisch (plötzliche und heftige Attacken, die weniger als eine Stunde dauern) oder kontinuierlich an den Zielorganen (z. B. Netzhautprobleme) auftreten.
Die Symptome eines Phäochromozytoms können sich plötzlich verschlimmern, was zu hypertensiven Krisen führt; dies geschieht hauptsächlich als Folge von emotionalem Stress oder Angst, chirurgischer Anästhesie oder körperlicher Anstrengung, die einen gewissen Druck auf den Tumor ausüben (Positionsänderungen, Sport, diagnostische Manöver (Katheterisierung), Gewichtheben, Stuhlgang, Wasserlassen, Schwangerschaft usw. ) . Die Symptome eines Phäomokrozytoms können auch durch die Einnahme bestimmter Medikamente (Amphetamine, hochdosiertes Koffein, Ephedrin, abschwellende Mittel, Antihistaminika, Kokain usw.) und Nahrungsmittel (tyraminreiche), die den Blutdruck erhöhen, verschlimmert werden Phäochromiktom sollte auch vermieden werden die Verwendung von Metoclopramid, Atropin und MAO-Hemmern (Risiko einer hypertensiven Krise mit Herzschäden); unter letzteren erinnern wir an Isocarboxazid, Phenelzin, Selegilin, Moclobemid und Tranylcypromin Hypertonische Krisen können mit Medikamenten wie Nitroglycerin und Derivaten kontrolliert werden.
Diagnose
Siehe auch: Metanephrine im Urin
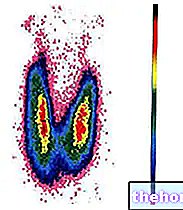
Die Diagnose eines Phäochromozytoms basiert auf der Dosierung von Adrenalin und Noradrenalin sowie deren Metaboliten (Metanephrine) im Blut und Urin. Bei erhöhten Spiegeln wird die genaue Lokalisation des Phäochromozytoms durch CT, abdominale MRT, 6-(18F)-Fluorodopamin-PET-Scan oder durch Meta-Iodbenzylguanidin (MIBG) oder Somatostatin-Analoga-Szintigraphie (Octreoscan) identifiziert.
Behandlung
Die Therapie der Wahl ist, wenn möglich, die chirurgische.
Mit der Entfernung der vom Phäochromozytom befallenen Nebenniere neigen die Symptome in den meisten Fällen zum Verschwinden, auch der Herzdruck normalisiert sich. Betreffen die Neoplasien beide Drüsen, müssen daher beide Nebennieren entfernt werden, leider erfordert die beidseitige Adrenalektomie eine chronische Ersatztherapie ersetzen verschiedene Nebennierenhormone wie Cortisol und Aldosteron.
Wenn immer möglich, wird laparoskopisch operiert, dh durch das Einführen feiner Präzisionsinstrumente in kleine Schnitte im Unterleib des Patienten.
Die Einnahme von Alpha- und Betablockern und ggf. anderen Medikamenten zur Kontrolle des Blutdrucks und der verschiedenen durch das Phäochromozytom ausgelösten Symptome ist während der Wartezeit und Vorbereitung auf die Operation unerlässlich.