Das Neurofibrom existiert in zwei verschiedenen Typen (dermal und plexiform) und tritt nach einer bestimmten Mutation des NF1-Gens auf; diese Mutation kann ohne ersichtlichen Grund oder als Folge einer als Typ-1-Neurofibromatose bekannten Erkrankung auftreten.
Neurofibrome unbekannter Ursache sind in der Regel einmalig, während Neurofibrome im Zusammenhang mit Neurofibromatose Typ 1 in der Regel mehrfach vorhanden sind (d. h. der Patient entwickelt mehrere Neurofibrome).
Neurofibrom ist ein Tumor, der behandelt wird, wenn die damit verbundenen Symptome schwerwiegend sind oder ein echtes Risiko für Komplikationen besteht.
Allgemeine Merkmale
Neurofibrom ist eine abnorme Formation, die neben der Nervenzellkomponente, aus der es stammt, auch enthält: Entzündungszellen (Mastzellen), Blutzellen und Bindegewebszellen.
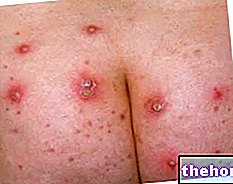
Als Einzel- oder Mehrfachläsion ist das Neurofibrom außerhalb des menschlichen Körpers weithin sichtbar, da es bei seinen Trägern eine Haut oder eine subkutane Ausstülpung von unterschiedlicher Größe und in einigen Fällen entstellend bestimmt.
Neurofibrom nach WHO
Nach dem System der WHO (World Health Organization) zur Klassifizierung von Tumoren des Nervensystems ist das Neurofibrom ein Grad-I-Tumor, d. h. ein gut differenzierter, langsam wachsender, nicht bösartiger Tumor mit langer Überlebenszeit (durch den Patienten) .
WHO-Klassifikation von Tumoren des zentralen und peripheren Nervensystems
Tumorgrad
Eigenschaften von Tumoren
Gut differenziert
Langsames Wachstum
Nicht bösartig
Verbunden mit langem ÜberlebenMäßig differenziert
Relativ langsames Wachstum
Bösartig oder nicht bösartig
Sie können sich zu höhergradigen Tumoren entwickelnDifferenziert
Schnelles Wachstum
Maligne
Sie können sich zu höhergradigen Tumoren entwickeln
Verbunden mit geringer ÜberlebenschanceHoch differenziert
Schnelles Wachstum
Maligne
Verbunden mit geringer ÜberlebenschanceUnter Vorbehalt der Details für die nächsten Abschnitte dieses Kapitels gehören alle Neurofibrome, die mit einem einzelnen oberflächlichen peripheren Nerv assoziiert sind, zum dermalen Typ, während alle Neurofibrome, die mit einem Komplex oberflächlicher peripherer Nerven assoziiert sind, und alle Neurofibrome, die mit einem Komplex oberflächlicher Nerven assoziiert sind, zu den plexiforme Art. tiefe periphere Nerven.
Dermale Neurofibrom
Es gibt zwei Subtypen des dermalen Neurofibroms: den diskreten kutanen Subtyp (oder diskreten kutanen Neurofibrom) und den diskreten subkutanen Subtyp (oder diskreten subkutanen Neurofibrom).
- Merkmale des diskreten kutanen Neurofibroms: es handelt sich um einen Hautvorsprung (er befindet sich auf der Hautoberfläche); es kann gestielt oder nicht gestielt sein; es ist fleischig; es hat keine zarte Konsistenz; es ist variabel in der Größe.
- Merkmale des diskreten subkutanen Neurofibroms: auf der Haut erscheint sie als subkutane Ausstülpung (sie befindet sich in den ersten Hautschichten); es kann sich weich anfühlen.
Plexiformes Neurofibrom
Plexiformes Neurofibrom ist eine große Tumormasse; Aus diesem Grund betrifft es eher oberflächliche oder tiefe periphere Nerven.
Wenn es in die Nähe eines Komplexes tiefer peripherer Nerven wächst, befindet sich das plexiforme Neurofibrom unterhalb der Dermis und kann nicht nur in den betroffenen Nervenstrukturen, sondern auch in den an diese Nervenstrukturen angrenzenden Organen zum Protagonisten eines Kompressionsprozesses werden.
Wie im vorherigen Fall gibt es zwei Subtypen des plexiformen Neurofibroms: den diffusen Subtyp (oder diffuses plexiformes Neurofibrom) und den nodulären Subtyp (oder noduläres plexiformes Neurofibrom).
- Merkmale des diffusen plexiformen Neurofibroms: erscheint als subkutane Schwellung von weicher Konsistenz; die Ränder sind schlecht definiert.
- Merkmale des nodulären plexiformen Neurofibroms: er hat seinen Sitz unterhalb der Dermis, bestimmt aber trotzdem eine von außen sichtbare Ausstülpung; er kann ei- oder kugelförmig sein; die Konsistenz ist hart; er ist gut umschrieben.
Plexiforme Neurofibrome sind nie eine „Evolution dermaler Neurofibrome, sondern Formationen, die als solche entstehen.

Die Neurofibromatose Typ 1 ist ein Beispiel für eine autosomal-dominant vererbte Erkrankung.
Andere Anzeichen und Symptome einer Neurofibromatose Typ 1
Die Neurofibromatose Typ 1 ist für ein reichhaltiges Symptombild verantwortlich; neben Neurofibrom verursacht es tatsächlich: kaffee- und milchfarbene Hautflecken, gutartige Hauttumore, gutartige Tumoren der Iris, gutartige Hirntumore, physikalisch-skelettale Anomalien, Sehstörungen und Bluthochdruck.
Welche Funktionen hat das unveränderte NF1? Was passiert stattdessen, wenn es sich geändert hat?
Prämisse: Die auf menschlichen Chromosomen vorhandenen Gene sind DNA-Sequenzen, die die Aufgabe haben, grundlegende Proteine für lebenswichtige biologische Prozesse, einschließlich Zellwachstum und -replikation, zu produzieren.
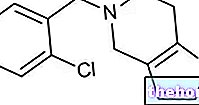
Im gesunden (d. h. frei von Mutationen) Gen produziert das NF1-Gen ein Protein namens Neurofibromin, das für die Regulierung des Wachstumsprozesses von Nervenzellen verantwortlich ist.
Auf der anderen Seite verliert NF1, wenn es die für Neurofibrome verantwortliche Mutation trägt, die Fähigkeit, Neurofibromin zu bilden, und dies führt zu einer völlig abnormalen Entwicklung und einem abnormalen Wachstum von Nervenzellen.
Wussten Sie, dass ...
Das NF1-Gen ist eines der am meisten mutierten Gene im menschlichen Genom; diese bleiben jedoch oft ohne Folgen, da das betreffende Gen sehr groß ist und zahlreiche DNA-Anteile enthält, die für die endgültige Bildung des Neurofibromin-Proteins keine besondere Bedeutung haben.
Welche Zellen des peripheren Nervensystems sind davon betroffen?
Innerhalb des peripheren Nervensystems betrifft Neurofibrom nicht-myelinisierte Schwann-Zellen, die myelinfreie Version der bekannteren myelinisierten Schwann-Zellen.
Für Neurofibrome sind nicht-myelinisierte Schwann-Zellen hochspezifische Ziele; obwohl sie zum gleichen Zelltyp gehören wie myelinisierte Schwann-Zellen, sind nur sie von der NF1-Mutation betroffen, die für den Entstehungsprozess des Neurofibroms verantwortlich ist.
Der Grund, warum Neurofibrome mit derselben NF1-Mutation nur aus nicht-myelinisierten Schwann-Zellen stammen, ist völlig unbekannt und stellt eines der am meisten untersuchten Themen von Genetikern und Molekularbiologie-Experten dar.
Epidemiologie
Laut Statistik sind Neurofibrome in 90% der Fälle idiopathisch und nur in den verbleibenden 10% der Fälle auf das Vorliegen einer Typ-1-Neurofibromatose zurückzuführen; dennoch verdanken sie letzteren den größten Teil ihrer Bekanntheit.
EPIDEMIOLOGIE DER NEUROFIBROMATOSE TYP 1
Statistiken zeigen, dass eines von 3.000 Neugeborenen Träger einer Typ-1-Neurofibromatose ist; sie bezeugen auch, dass die fragliche genetische Krankheit in 50 % der Fälle erblich ist und in den restlichen 50 % der Fälle während der Embryonalentwicklung erworben wurde (daher besteht eine Äquivalenz zwischen der Anzahl der erblichen Fälle und der Anzahl der nicht erblichen Fälle).
, Brennen und Juckreiz. Andererseits verursacht das Neurofibrom, wenn es plexiform ist, Anzeichen von Hypertrophie, Hyperpigmentierung und Hypertrichose auf der darüber liegenden Haut; Darüber hinaus kann es, wenn es nahe an tiefen peripheren Nerven wächst, zu neurologischen Kompressionsproblemen führen.Dermale Neurofibrom und Typ-1-Neurofibromatose
Bei Menschen mit Typ-1-Neurofibromatose tritt das dermale Neurofibrom im Allgemeinen in Form von multiplen Läsionen auf, beginnend mit der Adoleszenz (die Pubertät ist oft ein kritischer Moment, da sie den Beginn der ersten Neurofibrome markiert); im Laufe der Jahre steigt daher die Zahl und die Größe dieser Läsionen nimmt progressiv zu (NB bei Größe c "ist eine Grenze, in dem Sinne, dass sie ab einer bestimmten Größe nicht weiter wachsen) und erreichen einen großen Teil des Körpers des Patienten.
Plexiformes Neurofibrom und Typ-1-Neurofibromatose
Bei Patienten mit Neurofibromatose Typ 1 tritt das plexiforme Neurofibrom auch in Form von multiplen Läsionen in sehr jungen Jahren auf (also vor dermalen Neurofibromen); unter den fraglichen Umständen sind die am stärksten betroffenen anatomischen Stellen der Augapfel, das Gesicht, Arme, Beine und Rumpf.
Komplikationen des dermalen Neurofibroms
Dermale Neurofibrom verursacht Komplikationen, wenn er in Form von multiplen Läsionen im Rahmen einer Typ-1-Neurofibromatose auftritt; unter solchen Umständen kann es sogar das Aussehen des Patienten auf eine entschieden tiefgreifende Weise entstellen.
KANN ES ZU EINEM BÖSEN KREBS ENTWICKELN?
Bis heute hat kein Mitglied der medizinisch-wissenschaftlichen Gemeinschaft Fälle von dermalen Neurofibromen registriert, die sich zu Krebserkrankungen bösartiger Natur entwickeln. Dies deutet darauf hin, dass es unmöglich ist, dass aus dem dermalen Neurofibrom ein bösartiges Neoplasma entsteht.
Komplikationen des plexiformen Neurofibroms
Das plexiforme Neurofibrom ist entschieden gefährlicher als das dermale Neurofibrom; tatsächlich können verschiedene Komplikationen, einige sogar sehr schwerwiegend, von seinem Vorhandensein abhängen.
Zu den möglichen Komplikationen gehören:
- Die Möglichkeit, das Erscheinungsbild des Patienten zu entstellen, auch wenn die Präsentationsmodalität einmalig ist;
- Die Möglichkeit, nach einer Kompression der betroffenen Nerven schwere neurologische Ausfälle zu verursachen;
- Die Möglichkeit, sich zu einem bösartigen Tumor zu entwickeln.
BÖSE EVOLUTION EINES PLEXIFORMEN NEUROPHIBROMS: DIE DETAILS
Plexiformes Neurofibrom kann sich zu einem bösartigen Tumor entwickeln, der als bösartiger Tumor der peripheren Nervenscheiden (MPNST) bekannt ist.
Laut statistischen Studien würde der Prozess der malignen Evolution, dessen Protagonist das plexiforme Neurofibrom werden kann, etwa 9-10% der klinischen Fälle betreffen.
- Ein CT-Scan oder eine kernmagnetische Resonanz der anatomischen Stelle, an der sich der/die Tumor(e) befinden, um die Lage und das Ausmaß der Tumore zu untersuchen;
- Eine Tumorbiopsie zur diagnostischen Bestätigung von Neurofibromen;
- Ein genetischer Test, um festzustellen, ob der Patient eine Neurofibromatose vom Typ 1 hat.
Behandlung eines plexiformen Neurofibroms, das zu einem bösartigen Tumor geworden ist
Die maligne Entwicklung eines plexiformen Neurofibroms erfordert immer den Einsatz einer Behandlung zur Entfernung der Tumormasse.
Mögliche Behandlungen für einen bösartigen Tumor der peripheren Nervenscheiden sind: Operation, Strahlentherapie und Chemotherapie.
Die postoperative Phase nach der Entfernung des plexiformen Neurofibroms
Nach einer Operation zur Entfernung plexiformer Neurofibrome oder ihrer malignen Versionen muss der Patient eine Reihe von physiotherapeutischen Rehabilitationsbehandlungen durchlaufen, um sich von bestimmten neurologischen Defiziten, die durch die Nervenresektion verursacht wurden, zu erholen.
Mögliche Behandlungen der Zukunft
Kürzlich haben Experten, die sich mit der Erforschung neuer Therapien gegen Neurofibrome beschäftigen, festgestellt, dass letztere bei Exposition mit ACE-Hemmern eine Verlangsamung der Entwicklung zu erfahren scheinen; ACE-Hemmer sind, wie vielen Lesern vielleicht bereits bekannt, Medikamente zur Behandlung von Bluthochdruck und andere Herz-Kreislauf-Erkrankungen.
, von denen Metastasen abhängen können).In einem solchen Kontext hat die Art der Präsentation eine relative Bedeutung in dem Sinne, dass die mit dem plexiformen Neurofibrom verbundenen Gefahren sowohl bestehen, wenn letzteres in einer einzigen Form auftritt, als auch wenn es in mehreren Formen auftritt.