Wirkstoffe: Erlotinib
Tarceva 25 mg Filmtabletten
Tarceva 100 mg Filmtabletten
Tarceva 150 mg Filmtabletten
Warum wird Tarceva verwendet? Wofür ist das?
Tarceva enthält den Wirkstoff Erlotinib. Tarceva ist ein Arzneimittel zur Behandlung von Krebs und wirkt, indem es die Aktivität eines Proteins namens epidermaler Wachstumsfaktorrezeptor (EGFR) blockiert, das am Wachstum und der Ausbreitung von Krebszellen beteiligt ist.
Tarceva ist für Erwachsene indiziert. Dieses Arzneimittel kann Ihnen verschrieben werden, wenn Sie an fortgeschrittenem nicht-kleinzelligem Lungenkrebs leiden. Es kann Ihnen als Ersttherapie oder als Therapie verordnet werden, wenn die Erkrankung nach der initialen Chemotherapie weitgehend unverändert bleibt, sofern die Krebszellen spezifische EGFR-Mutationen aufweisen, oder auch, wenn eine vorangegangene Chemotherapie den Krebs nicht stoppen konnte .Krankheit.
Dieses Arzneimittel kann Ihnen auch in Kombination mit einer anderen Behandlung namens Gemcitabin verschrieben werden, wenn Sie an metastasierendem Bauchspeicheldrüsenkrebs leiden.
Kontraindikationen Wann Tarceva nicht angewendet werden sollte
Tarceva® darf nicht eingenommen werden
- wenn Sie allergisch gegen Erlotinib oder einen der in Abschnitt 6. genannten sonstigen Bestandteile dieses Arzneimittels sind.
Vorsichtsmaßnahmen für die Anwendung Was sollten Sie vor der Einnahme von Tarceva® beachten?
Warnungen und Vorsichtsmaßnahmen:
- wenn Sie andere Arzneimittel einnehmen, die die Menge von Erlotinib in Ihrem Blut erhöhen oder erniedrigen oder seine Wirksamkeit beeinträchtigen können (z. B. Antimykotika wie Ketoconazol, Proteasehemmer, Erythromycin, Clarithromycin, Phenytoin, Carbamazepin, Barbiturate, Rifampicin, Ciprofloxazolacin, Omepr Ranitidin, Johanniskraut oder Proteasom-Hemmer), fragen Sie Ihren Arzt. In einigen Fällen können diese Arzneimittel die Wirksamkeit von Tarceva verringern oder die Nebenwirkungen verstärken, und in diesem Fall muss Ihr Arzt möglicherweise Ihre Therapie anpassen.Ihr Arzt wird möglicherweise veranlassen, dass Sie diese Arzneimittel nicht einnehmen, während Sie mit Tarceva behandelt werden.
- wenn Sie Antikoagulanzien (Arzneimittel zur Vorbeugung von Thrombosen oder Blutgerinnseln, z. B. Warfarin) einnehmen, kann Tarceva die Blutungsneigung erhöhen. Sprechen Sie mit Ihrem Arzt, der Sie regelmäßig überwachen muss, indem er Ihnen einige Bluttests verordnet.
- wenn Sie Statine (Arzneimittel zur Senkung des Cholesterinspiegels im Blut) einnehmen, kann Tarceva das Risiko von statinbedingten Muskelproblemen erhöhen, die in seltenen Fällen zu einem schweren Muskelabbau (Rhabdomyolyse) mit Nierenschäden führen können. Sprechen Sie mit Ihrem Arzt.
- wenn Sie Kontaktlinsen verwenden und/oder jemals Augenprobleme wie schwere trockene Augen, Entzündungen der Augenvorderseite (Hornhaut) oder Geschwüre, die die Augenvorderseite befallen haben, hatten, sprechen Sie mit Ihrem Arzt. Siehe auch unter „Andere Arzneimittel und Tarceva“.
Sie müssen Ihren Arzt informieren:
- wenn Sie „plötzliche Atembeschwerden in Verbindung mit Husten oder Fieber haben, da Ihr Arzt möglicherweise andere Arzneimittel verschreiben und die Einnahme von Tarceva abbrechen muss;
- wenn Sie Durchfall haben, da Ihr Arzt möglicherweise Antidiarrhoika (z. B. Loperamid) verschreiben muss;
- wenn Sie schweren oder anhaltenden Durchfall, Übelkeit, Appetitlosigkeit oder Erbrechen haben, da Ihr Arzt Ihre Behandlung mit Tarceva möglicherweise abbrechen muss und Sie eine Behandlung im Krankenhaus benötigen.
- wenn Sie starke Bauchschmerzen, schwere Hautreaktionen wie Blasenbildung oder Abschälen haben.Ihr Arzt kann es für erforderlich halten, die Behandlung zu unterbrechen oder zu beenden.
- Wenn Sie eine akute rote oder sich verschlimmernde Augenrötung mit Schmerzen, vermehrtem Tränenfluss, verschwommenem Sehen und/oder Lichtempfindlichkeit entwickeln, sprechen Sie sofort mit Ihrem Arzt oder dem medizinischen Fachpersonal, da Sie möglicherweise eine dringende Behandlung benötigen (siehe „Welche Nebenwirkungen sind möglich?).
- wenn Sie zusätzlich ein Statin einnehmen und unerklärliche Muskelschmerzen, Empfindlichkeit, Schwäche oder Krämpfe verspüren. Der Arzt kann es für erforderlich halten, die Behandlung zu unterbrechen oder auszusetzen.
Siehe auch Abschnitt 4 „Mögliche Nebenwirkungen“.
Leber- und Nierenerkrankungen
Es ist nicht bekannt, ob sich die Wirkung von Tarceva ändert, wenn Leber oder Nieren nicht normal funktionieren. Wenn Sie eine schwere Leber- oder Nierenerkrankung haben, wird die Behandlung mit diesem Arzneimittel nicht empfohlen.
Glukuronierungsstörung wie das Gilbert-Syndrom
Wenn Sie an einer Glukuronidierungsstörung wie dem Gilbert-Syndrom leiden, sollte Ihr Arzt Sie mit Vorsicht behandeln.
Rauch
Wenn Sie Tarceva einnehmen, sollten Sie mit dem Rauchen aufhören, da Rauchen die Menge des Arzneimittels in Ihrem Blut verringern kann.
Kinder und Jugendliche
Tarceva wurde bei Patienten unter 18 Jahren nicht untersucht. Die Behandlung mit diesem Arzneimittel wird bei Kindern und Jugendlichen nicht empfohlen.
Wechselwirkungen Welche Medikamente oder Lebensmittel können die Wirkung von Tarceva® verändern?
Einnahme von Tarceva® zusammen mit anderen Arzneimitteln
Informieren Sie Ihren Arzt oder Apotheker, wenn Sie andere Arzneimittel einnehmen, kürzlich andere Arzneimittel eingenommen haben oder beabsichtigen andere Arzneimittel einzunehmen.
Einnahme von Tarceva zusammen mit Nahrungsmitteln und Getränken
Nehmen Sie Tarceva nicht zusammen mit Nahrung ein. Siehe auch Abschnitt 3 „Wie ist Tarceva einzunehmen“
Warnungen Es ist wichtig zu wissen, dass:
Schwangerschaft und Stillzeit
Vermeiden Sie eine Schwangerschaft während der Behandlung mit Tarceva. Wenn Sie vermuten, schwanger zu sein, wenden Sie während der Behandlung und mindestens 2 Wochen nach Einnahme der letzten Tablette eine angemessene Verhütungsmethode an.Wenn Sie während der Einnahme von Tarceva schwanger werden, informieren Sie sofort Ihren Arzt, der über die Fortsetzung der Behandlung entscheidet. Wenn Sie schwanger sind, oder wenn Sie vermuten, schwanger zu sein oder beabsichtigen, schwanger zu werden, oder wenn Sie stillen, fragen Sie vor der Einnahme dieses Arzneimittels Ihren Arzt oder Apotheker um Rat.
Verkehrstüchtigkeit und das Bedienen von Maschinen
Es wurden keine Studien zu den möglichen Auswirkungen von Tarceva auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen durchgeführt, aber es ist sehr unwahrscheinlich, dass die Behandlung diese Fähigkeit verändert.
Überempfindlichkeit
Tarceva enthält einen Zucker namens Lactose-Monohydrat. Bitte nehmen Sie Tarceva erst nach Rücksprache mit Ihrem Arzt ein, wenn Ihnen bekannt ist, dass Sie unter einer Zuckerunverträglichkeit leiden.
Dosis, Methode und Zeitpunkt der Anwendung Wie ist Tarceva anzuwenden: Dosierung
Nehmen Sie dieses Arzneimittel immer genau nach Absprache mit Ihrem Arzt ein. Fragen Sie im Zweifelsfall Ihren Arzt oder Apotheker.
Die Tablette sollte mindestens eine Stunde vor oder zwei Stunden nach dem Essen eingenommen werden.
Die übliche Dosis beträgt eine Tablette Tarceva 150 mg täglich, wenn Sie an nicht-kleinzelligem Lungenkrebs leiden. Die übliche Dosis beträgt eine Tablette Tarceva 100 mg täglich, wenn Sie an metastasierendem Bauchspeicheldrüsenkrebs leiden. Tarceva wird in Kombination mit Gemcitabin verabreicht.
Ihr Arzt kann die Dosis um jeweils 50 mg ändern. Für die verschiedenen Dosierungsschemata ist Tarceva in den Stärken 25 mg, 100 mg oder 150 mg erhältlich.
Überdosierung Was ist zu tun, wenn Sie zu viel Tarceva eingenommen haben?
Wenn Sie eine größere Menge von Tarceva eingenommen haben, als Sie sollten
Wenden Sie sich umgehend an Ihren Arzt oder Apotheker. Es kann sein, dass sich die Nebenwirkungen verschlimmern und Ihr Arzt Sie auffordern wird, die Einnahme abzubrechen.
Wenn Sie die Einnahme von Tarceva vergessen haben
Wenn Sie eine oder mehrere Dosen von Tarceva vergessen haben, wenden Sie sich so schnell wie möglich an Ihren Arzt oder Apotheker. Nehmen Sie nicht die doppelte Dosis ein, wenn Sie die vorherige Einnahme vergessen haben.
Wenn Sie die Einnahme von Tarceva® abbrechen
Es ist wichtig, Tarceva jeden Tag so lange einzunehmen, wie Ihr Arzt es Ihnen verordnet.
Wenn Sie weitere Fragen zur Anwendung dieses Arzneimittels haben, wenden Sie sich an Ihren Arzt oder Apotheker.
Nebenwirkungen Was sind die Nebenwirkungen von Tarceva
Wie alle Arzneimittel kann auch dieses Arzneimittel Nebenwirkungen haben, die aber nicht bei jedem auftreten müssen.
Wenn bei Ihnen eine der unten aufgeführten Nebenwirkungen auftritt, wenden Sie sich bitte so schnell wie möglich an Ihren Arzt. In einigen Fällen muss der Arzt möglicherweise die Dosis von Tarceva reduzieren oder die Behandlung abbrechen.
- Durchfall und Erbrechen (sehr häufig, kann mehr als 1 von 10 Patienten betreffen). Anhaltender und schwerer Durchfall kann zu einer verminderten Kaliumkonzentration im Blut und zu Nierenversagen führen, insbesondere wenn Sie gleichzeitig mit anderen Chemotherapeutika behandelt werden. Bei schwererem oder anhaltendem Durchfall wenden Sie sich sofort an Ihren Arzt, der sich möglicherweise für eine stationäre Behandlung entscheidet.
- Augenreizung aufgrund von Konjunktivitis/Keratokonjunktivitis (sehr häufig, kann mehr als 1 von 10 Patienten betreffen) und Keratitis (häufig, kann bis zu 1 von 10 Patienten betreffen).
- Eine Form der Lungenentzündung, die als interstitielle Lungenerkrankung bezeichnet wird (gelegentlich bei europäischen Patienten, häufig bei japanischen Patienten: kann bis zu 1 von 100 Patienten in Europa und bis zu 1 von 10 in Japan betreffen). Diese Krankheit kann auch mit dem natürlichen Fortschreiten Ihrer Erkrankung in Verbindung gebracht werden und in einigen Fällen tödlich sein. Wenn bei Ihnen Symptome wie „plötzliche Atembeschwerden in Verbindung mit Husten oder Fieber“ auftreten, wenden Sie sich sofort an Ihren Arzt, da Sie möglicherweise an dieser Krankheit leiden. Ihr Arzt wird möglicherweise entscheiden, die Behandlung mit Tarceva dauerhaft zu beenden.“
- Es wurden Fälle von Magen-Darm-Perforation beobachtet (gelegentlich, kann bis zu 1 von 100 Patienten betreffen). Informieren Sie Ihren Arzt, wenn Sie starke Bauchschmerzen haben oder wenn Sie jemals ein Magengeschwür oder eine Divertikelerkrankung hatten, da dies das Perforationsrisiko erhöhen kann.
- In seltenen Fällen wurde Leberversagen beobachtet (selten, kann bis zu 1 von 1000 Behandelten betreffen.) Wenn Ihre Blutuntersuchungen auf schwere Veränderungen der Leberfunktion hinweisen, kann Ihr Arzt entscheiden, die Behandlung abzubrechen.
Sehr häufige Nebenwirkungen (kann mehr als 1 von 10 Patienten betreffen):
- Hautausschlag, der sich in sonnenexponierten Bereichen entwickeln oder verschlimmern kann. Wenn Sie der Sonne ausgesetzt sind, kann es ratsam sein, Schutzkleidung und/oder Sonnenschutzmittel (z. B. auf Basis von Mineralstoffen) zu verwenden.
- Infektion
- Appetitlosigkeit, Gewichtsverlust
- Depression
- Kopfschmerzen, Veränderungen des Hautgefühls oder Taubheitsgefühl in den Extremitäten
- Atembeschwerden, Husten
- Brechreiz
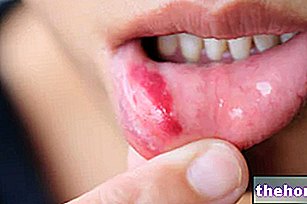
- Reizungen im Mund
- Magenschmerzen, Verdauungsstörungen und Blähungen
- Veränderungen bei Bluttests im Zusammenhang mit der Leberfunktion
- Juckende, trockene Haut und Haarausfall
- Müdigkeit, Fieber, Schüttelfrost
Häufige Nebenwirkungen (kann bis zu 1 von 10 Patienten betreffen):
- Nasenbluten
- Blutungen aus Magen oder Darm
- Entzündungsreaktionen um die Nägel
- Infektion der Haarfollikel
- Akne
- Spalthaut (Hautrisse)
- Verminderte Nierenfunktion (bei Anwendung außerhalb der zugelassenen Indikationen in Kombination mit Chemotherapie)
Gelegentliche Nebenwirkungen (kann bis zu 1 von 100 Behandelten betreffen)
- Veränderungen der Wimpern
- übermäßige Behaarung im Gesicht und am Körper mit männlicher Musterverteilung
- Veränderungen der Augenbrauen
- brüchige und abblätternde Nägel
Seltene Nebenwirkungen (kann bis zu 1 von 1000 Patienten betreffen):
- Rötung oder Schmerzen in den Handflächen oder Fußsohlen (palmar-plantares Erythrodysästhesie-Syndrom)
Sehr seltene Nebenwirkungen (kann bis zu 1 von 10.000 Behandelten betreffen)
- Fälle von Hornhautulzeration oder -perforation
- Schwere Hautreaktionen wie Blasenbildung oder Abschälen (Hinweis auf Stevens-Johnson-Syndrom)
- Entzündung des farbigen Teils des Auges
Meldung von Nebenwirkungen
Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt oder Apotheker, einschließlich aller möglichen Nebenwirkungen, die nicht in dieser Packungsbeilage aufgeführt sind. Sie können Nebenwirkungen auch direkt über das in Anhang V aufgeführte nationale Meldesystem melden. Indem Sie Nebenwirkungen melden, können Sie dazu beitragen, dass mehr Informationen über die Sicherheit dieses Arzneimittels zur Verfügung gestellt werden.
Ablauf und Aufbewahrung
Bewahren Sie dieses Arzneimittel für Kinder unzugänglich auf.
Verwenden Sie dieses Arzneimittel nicht nach dem Verfallsdatum, das auf der Blisterpackung und dem Karton nach EXP / EXP angegeben ist.Das Verfallsdatum bezieht sich auf den letzten Tag des Monats.
Für dieses Arzneimittel sind keine besonderen Lagerungsbedingungen erforderlich.
Werfen Sie Arzneimittel nicht in das Abwasser oder den Hausmüll. Fragen Sie Ihren Apotheker, wie Sie Arzneimittel, die Sie nicht mehr verwenden, entsorgen. Dies trägt zum Schutz der Umwelt bei.
Zusammensetzung und Darreichungsform
Was Tarceva enthält:
- Der Wirkstoff in Tarceva ist Erlotinib. Jede Filmtablette enthält je nach Stärke 25 mg, 100 mg oder 150 mg Erlotinib (als Erlotinibhydrochlorid).
- Die sonstigen Bestandteile sind: Tablettenkern: Lactose-Monohydrat, mikrokristalline Cellulose, Natriumstärkeglycolat Typ A, Natriumdodecylsulfat, Magnesiumstearat (siehe auch Abschnitt 2 zu Lactose-Monohydrat). Tablettenüberzug: Hypromellose, Hydroxypropylcellulose, Titandioxid, Macrogol.
Wie Tarceva aussieht und Inhalt der Packung:
Tarceva 25 mg wird als weiße bis gelbliche, runde Filmtablette mit der Prägung „T 25“ auf einer Seite geliefert und ist in Packungen mit 30 Tabletten erhältlich.
Tarceva 100 mg wird als weiße bis gelbliche, runde Filmtablette mit der Prägung „T 100“ auf einer Seite geliefert und ist in Packungen mit 30 Tabletten erhältlich.
Tarceva 150 mg wird als weiße bis gelbliche, runde Filmtablette mit der Prägung „T 150“ auf einer Seite geliefert und ist in Packungen mit 30 Tabletten erhältlich.
Quelle Packungsbeilage: AIFA (Italienische Arzneimittelbehörde). Im Januar 2016 veröffentlichter Inhalt. Die vorliegenden Informationen können nicht aktuell sein.
Um Zugriff auf die aktuellste Version zu erhalten, ist es ratsam, auf die Website der AIFA (Italienische Arzneimittelbehörde) zuzugreifen. Haftungsausschluss und nützliche Informationen.
01.0 BEZEICHNUNG DES ARZNEIMITTELS
TARCEVA 150 MG TABLETTEN MIT FILM . BESCHICHTET
02.0 QUALITATIVE UND QUANTITATIVE ZUSAMMENSETZUNG
Eine Filmtablette enthält 150 mg Erlotinib (als Erlotinib-Hydrochlorid).
Sonstige Bestandteile mit bekannter Wirkung: Jede Filmtablette enthält 103,82 mg Lactose-Monohydrat.
Die vollständige Auflistung der sonstigen Bestandteile finden Sie in Abschnitt 6.1.
03.0 DARREICHUNGSFORM
Filmtablette.
Weiße bis gelbliche, runde, bikonvexe Tabletten mit der Prägung „T 150“ auf einer Seite.
04.0 KLINISCHE INFORMATIONEN
04.1 Anwendungsgebiete
Nicht-kleinzelliges Lungenkarzinom ( Nicht-kleinzelligem Lungenkrebs , NSCLC):
Tarceva ist angezeigt zur Erstlinienbehandlung von Patienten mit lokal fortgeschrittenem oder metastasiertem nicht-kleinzelligem Lungenkrebs (NSCLC) mit aktivierenden EGFR-Mutationen.
Tarceva ist auch als Switch-Erhaltungstherapie bei Patienten mit lokal fortgeschrittenem oder metastasiertem NSCLC mit aktivierenden EGFR-Mutationen und stabiler Erkrankung nach Erstlinien-Chemotherapie angezeigt.
Tarceva ist auch angezeigt für die Behandlung von Patienten mit lokal fortgeschrittenem oder metastasiertem NSCLC nach Versagen mindestens einer vorherigen Chemotherapie.
Bei der Verschreibung von Tarceva sollten Faktoren berücksichtigt werden, die mit einer verlängerten Überlebenszeit verbunden sind.
Die Behandlung zeigte keinen Überlebensvorteil oder andere klinisch relevante Wirkungen bei Patienten mit epidermalen Wachstumsfaktorrezeptor (EGFR)-IHC-negativen Tumoren (siehe Abschnitt 5.1).
Bauchspeicheldrüsenkrebs:
Tarceva in Kombination mit Gemcitabin ist angezeigt zur Behandlung von Patienten mit metastasierendem Bauchspeicheldrüsenkrebs.
Bei der Verschreibung von Tarceva sollten Faktoren berücksichtigt werden, die mit einer verlängerten Überlebenszeit verbunden sind (siehe Abschnitte 4.2 und 5.1).
Für Patienten mit lokal fortgeschrittener Erkrankung wurde kein Überlebensvorteil nachgewiesen.
04.2 Dosierung und Art der Anwendung
Die Behandlung mit Tarceva sollte von einem Arzt überwacht werden, der Erfahrung in der Anwendung antineoplastischer Therapien hat.
Patienten mit nicht-kleinzelligem Lungenkarzinom:
Bei chemonaiven Patienten mit fortgeschrittenem oder metastasiertem NSCLC sollte vor Beginn der Tarceva-Therapie ein EGFR-Mutationstest durchgeführt werden.
Die empfohlene Tagesdosis von Tarceva beträgt 150 mg und wird mindestens eine Stunde vor oder zwei Stunden nach dem Essen eingenommen.
Patienten mit Bauchspeicheldrüsenkrebs:
Die empfohlene Tagesdosis von Tarceva beträgt 100 mg, die mindestens eine Stunde vor oder zwei Stunden nach dem Essen in Kombination mit Gemcitabin eingenommen wird (siehe Fachinformation von Gemcitabin in der Indikation Bauchspeicheldrüsenkrebs).
Bei Patienten, die in den ersten 4-8 Wochen der Therapie keinen Hautausschlag entwickeln, sollte erneut geprüft werden, ob die Behandlung mit Tarceva fortgesetzt werden soll (siehe Abschnitt 5.1).
Wenn die Dosis geändert werden muss, sollte sie jedes Mal um 50 mg reduziert werden (siehe Abschnitt 4.4).
Tarceva ist in den Stärken 25 mg, 100 mg und 150 mg erhältlich.
Die gleichzeitige Anwendung von CYP3A4-Substraten und -Modulatoren kann eine Änderung der Dosierung erforderlich machen (siehe Abschnitt 4.5).
Patienten mit Leberinsuffizienz: Die Elimination von Erlotinib erfolgt über den hepatischen Metabolismus und die biliäre Ausscheidung. Obwohl die Erlotinib-Exposition bei Patienten mit mäßiger Leberfunktionsstörung (Child-Pugh-Score von 7-9) und bei Patienten mit ausreichender Leberfunktion ähnlich war, ist bei der Anwendung von Tarceva bei Patienten mit Leberfunktionsstörung Vorsicht geboten eine Reduzierung oder ein Absetzen der Behandlung mit Tarceva sollte in Betracht gezogen werden. Die Sicherheit und Wirksamkeit von Erlotinib wurden bei Patienten mit schwerer Leberfunktionsstörung (AST / SGOT und ALT / SGPT > 5 x ULN) nicht untersucht. Tarceva wird für die Anwendung bei Patienten mit schwerer Leberfunktionsstörung nicht empfohlen (siehe Abschnitt 5.2).
Patienten mit Niereninsuffizienz: Die Sicherheit und Wirksamkeit von Erlotinib wurden bei Patienten mit Niereninsuffizienz (Serumkreatinin > 1,5-facher oberer Normwert) nicht untersucht. Basierend auf pharmakokinetischen Daten ist es bei Patienten mit leichter oder mittelschwerer Niereninsuffizienz nicht erforderlich, die Dosierung zu ändern ( siehe Abschnitt 5.2) Die Anwendung von Tarceva bei Patienten mit schwerer Niereninsuffizienz wird nicht empfohlen.
Kinder und Jugendliche: Die Sicherheit und Wirksamkeit von Erlotinib bei Patienten unter 18 Jahren ist nicht erwiesen Die Anwendung von Tarceva bei pädiatrischen Patienten wird nicht empfohlen.
Raucher: Zigarettenrauchen senkt die Erlotinib-Exposition nachweislich um 50-60 %. Die maximal verträgliche Dosis von Tarceva bei NSCLC-Patienten, die Zigaretten rauchen, betrug 300 mg. Eine langfristige Wirksamkeit und Sicherheit über der empfohlenen Anfangsdosis wurde bei Patienten nicht bestimmt die weiterhin rauchen (siehe Abschnitte 4.5 und 5.2). Daher sollte Rauchern geraten werden, mit dem Rauchen aufzuhören, da die Plasmakonzentrationen von Erlotinib bei Rauchern niedriger sind als bei Nichtrauchern.
04.3 Kontraindikationen
Überempfindlichkeit gegen Erlotinib oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
04.4 Besondere Warnhinweise und geeignete Vorsichtsmaßnahmen für die Anwendung
Beurteilung des EGFR-Mutationsstatus:
Um den EGFR-Mutationsstatus eines Patienten zu beurteilen, ist es wichtig, dass eine gut validierte und robuste Methodik gewählt wird, um falsch negative oder falsch positive Bestimmungen zu vermeiden.
Raucher
Rauchern sollte geraten werden, mit dem Rauchen aufzuhören, da die Plasmakonzentrationen von Erlotinib bei Rauchern niedriger sind als bei Nichtrauchern. Der Grad der Reduktion könnte klinisch signifikant sein (siehe Abschnitt 4.5).
Lungeninterstitielle Erkrankung (ILD .))
Gelegentlich wurden bei Patienten, die Tarceva zur Behandlung von nicht-kleinzelligem Lungenkrebs (NSCLC), Bauchspeicheldrüsenkrebs oder anderen fortgeschrittenen soliden Tumoren einnahmen, Fälle von ILD-ähnlichen Ereignissen, die manchmal tödlich waren, berichtet. In der Zulassungsstudie BR.21 bei NSCLC war die Inzidenz von ILD (0,8 %) in der Placebo- und in der Tarceva-Gruppe identisch, in der Pankreaskarzinom-Studie in Verbindung mit Gemcitabin betrug die Inzidenz ILD-ähnlicher Ereignisse 2,5 % in der Tarceva- und Gemcitabin-Gruppe im Vergleich zu 0,4 % in der Placebo- und Gemcitabin-Gruppe. Die Gesamtinzidenz bei mit Tarceva behandelten Patienten in allen Studien (einschließlich unkontrollierter und Patienten mit gleichzeitiger Chemotherapie) beträgt ungefähr 0,6 %, verglichen mit 0,2 % bei mit Placebo behandelten Patienten Lungenentzündung, Strahlenpneumonie, Überempfindlichkeitspneumonitis, interstitielle Pneumonie, interstitielle Lungenerkrankung, Bronchiolitis obliterans, Lungenfibrose, akutes Atemnotsyndrom, Alveolitis und "pulmonale Infiltration". Symptome traten innerhalb weniger Tage bis mehrere Monate nach Beginn der Tarceva-Therapie auf Störende oder gleichzeitige Faktoren wie begleitende oder frühere Chemotherapie, frühere Strahlentherapie, vorbestehende parenchymale Lungenerkrankung, Metastasen oder Lungeninfektionen Eine höhere Inzidenz von ILD (ca eine Sterblichkeitsrate von 1,5%) wurde bei der Bevölkerung japanischer Herkunft beobachtet.
Bei Patienten mit akuten neuen und/oder fortschreitenden ungeklärten Lungensymptomen wie Dyspnoe, Husten und Fieber sollte Tarceva bis zur diagnostischen Abklärung abgesetzt werden. Patienten, die gleichzeitig mit Erlotinib und Gemcitabin behandelt werden, sollten engmaschig auf die Möglichkeit einer ILD-ähnlichen Toxizität überwacht werden. Wenn eine ILD diagnostiziert wird, sollte Tarceva abgesetzt und nach Bedarf eine geeignete Behandlung eingeleitet werden (siehe Abschnitt 4.8).
Durchfall, Dehydration, Elektrolyt-Ungleichgewicht und Nierenversagen
Bei etwa 50 % der mit Tarceva behandelten Patienten trat Durchfall auf (einschließlich sehr seltener Fälle mit tödlichem Ausgang); mittelschwerer oder schwerer Durchfall sollte behandelt werden, zum Beispiel mit Loperamid. In einigen Fällen kann es erforderlich sein, die Dosis zu reduzieren. In klinischen Studien wurden die Dosen jeweils um 50 mg reduziert. Dosisreduktionen von jeweils 25 mg wurden nicht untersucht. Bei schwerem oder anhaltendem Durchfall, Übelkeit, Anorexie oder Erbrechen in Verbindung mit Dehydratation sollte die Anwendung von Tarceva abgebrochen und geeignete Maßnahmen zur Behandlung der Dehydratation ergriffen werden (siehe Abschnitt 4.8). Seltene Fälle von Hypokaliämie und Nierenversagen (einschließlich tödlicher Fälle) wurden berichtet. Einige Fälle waren sekundär zu einer schweren Dehydration, die durch Durchfall, Erbrechen und/oder Anorexie verursacht wurde, während andere auf eine begleitende Chemotherapie zurückzuführen waren.Bei schwererem oder anhaltendem Durchfall oder der zu Dehydration führt, insbesondere bei Patientengruppen mit erschwerenden Risikofaktoren (insbesondere in Verbindung mit einer Chemotherapie und anderen Medikamenten, Symptomen oder Krankheiten oder anderen prädisponierenden Zuständen, einschließlich im fortgeschrittenen Alter) sollte die Anwendung von Tarceva abgesetzt und geeignete Maßnahmen zur intensiven intravenösen Rehydratation des Patienten ergriffen werden. Darüber hinaus sollten bei Patienten, bei denen das Risiko einer Dehydratation besteht, die Nierenfunktion und die Serumelektrolyte, einschließlich Kalium, überwacht werden.
Hepatitis, Leberversagen
Während der Behandlung mit Tarceva wurden seltene Fälle von Leberversagen (einschließlich tödlicher Fälle) berichtet. Vorbestehende Lebererkrankungen oder die gleichzeitige Gabe von hepatotoxischen Arzneimitteln wurden als Störfaktoren angesehen. Bei solchen Patienten sollte daher eine regelmäßige Untersuchung der Leberfunktion in Betracht gezogen werden. Bei schweren Leberfunktionsstörungen sollte die Anwendung von Tarceva abgebrochen werden (siehe Abschnitt 4.8). Die Anwendung von Tarceva wird bei Patienten mit schwerer Leberfunktionsstörung nicht empfohlen.
Magen-Darm-Perforation
Patienten, die Tarceva einnehmen, haben ein erhöhtes Risiko, eine Magen-Darm-Perforation zu entwickeln, die gelegentlich (einschließlich einiger tödlicher Fälle) beobachtet wird. Das Risiko ist höher bei Patienten, die gleichzeitig antiangiogenetische Mittel, Kortikosteroide, NSAIDs und/oder eine Taxan-basierte Chemotherapie einnehmen, oder bei Patienten mit Magengeschwüren oder Divertikelerkrankungen in der Vorgeschichte. Bei Patienten, die eine Magen-Darm-Perforation entwickeln, sollte die Behandlung mit Tarceva dauerhaft abgesetzt werden (siehe Abschnitt 4.8).
Bullöse, exfoliative Hauterkrankungen
Es wurde über bullöse, vesikuläre und exfoliative Hauterkrankungen berichtet, einschließlich sehr seltener Fälle, die auf ein Stevens-Johnson-Syndrom / toxische epidermale Nekrolyse hindeuten, die in einigen Fällen tödlich verliefen (siehe Abschnitt 4.8). Die Behandlung mit Tarceva sollte unterbrochen oder abgebrochen werden, wenn der Patient schwere bullöse, vesikuläre oder exfoliative Störungen entwickelt. Patienten mit bullösen und exfoliativen Hautveränderungen sollten auf Hautinfektionen untersucht und gemäß den lokalen Richtlinien behandelt werden.
Augenerkrankungen
Patienten mit Anzeichen oder Symptomen, die auf eine Keratitis hindeuten, wie akute Augenentzündung oder Verschlechterung des Auges, Tränenfluss, Photophobie, verschwommenes Sehen, Augenschmerzen und/oder rote Augen, sollten umgehend an einen Augenarzt überwiesen werden. Wenn die Diagnose einer ulzerativen Keratitis bestätigt wird, sollte die Behandlung mit Tarceva abgebrochen oder abgebrochen werden. Wenn eine Keratitis diagnostiziert wird, sollten Nutzen und Risiken einer fortgesetzten Behandlung sorgfältig abgewogen werden. Tarceva sollte bei Patienten mit einer Vorgeschichte von Keratitis, ulzerativer Keratitis oder schwerem Trockenen Auge mit Vorsicht angewendet werden. Die Verwendung von Kontaktlinsen ist auch ein Risikofaktor für Keratitis und Ulzerationen. Sehr seltene Fälle von Hornhautperforation oder -ulzeration wurden während der Anwendung von Tarceva berichtet (siehe Abschnitt 4.8).
Wechselwirkungen mit anderen Arzneimitteln
Starke CYP3A4-Induktoren können die Wirksamkeit von Erlotinib verringern, während starke CYP3A4-Inhibitoren zu einer erhöhten Toxizität führen können Eine gleichzeitige Behandlung mit dieser Art von Substanzen sollte vermieden werden (siehe Abschnitt 4.5).
Andere Formen der Interaktion
Erlotinib ist durch eine Abnahme der Löslichkeit bei pH-Werten über 5 gekennzeichnet. Arzneimittel, die den pH-Wert des oberen Gastrointestinaltrakts (GI) verändern, wie Protonenpumpenhemmer, H2-Antagonisten und Antazida, können die Löslichkeit von Erlotinib beeinflussen und daher seine Bioverfügbarkeit. Eine Erhöhung der Tarceva-Dosis bei gleichzeitiger Anwendung mit diesen Arzneimitteln kann die Verringerung der Exposition möglicherweise nicht ausgleichen. Die Kombination von Erlotinib mit Protonenpumpenhemmern sollte vermieden werden. Die Auswirkungen einer gleichzeitigen Anwendung von Erlotinib mit H2-Antagonisten und Antazida sind nicht bekannt, jedoch besteht wahrscheinlich eine verringerte Bioverfügbarkeit. Die gleichzeitige Anwendung dieser sollte daher vermieden werden Abschnitt 4.5) Wenn während der Behandlung mit Tarceva Antazida als notwendig erachtet werden, sollten diese mindestens 4 Stunden vor oder 2 Stunden nach der Tagesdosis von Tarceva eingenommen werden.
Die Tabletten enthalten Lactose und sollten nicht an Patienten mit der seltenen hereditären Galactose-Intoleranz, Lapp-Lactase-Mangel oder Glucose-Galactose-Malabsorption verabreicht werden.
04.5 Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen
Interaktionsstudien wurden nur bei Erwachsenen durchgeführt.
Erlotinib und andere CYP-Substrate
Erlotinib ist ein starker Inhibitor von CYP1A1 und ein mäßiger Inhibitor von CYP3A4 und CYP2C8 sowie ein starker Inhibitor der Glucuronidierung in vitro durch die UGT1A1.
Aufgrund der stark reduzierten Expression von CYP1A1 in menschlichen Geweben ist die physiologische Relevanz der starken Hemmung von CYP1A1 nicht bekannt.
Bei gleichzeitiger Anwendung von Erlotinib mit Ciprofloxacin, einem mäßigen CYP1A2-Inhibitor, war die Erlotinib-Exposition [AUC] signifikant um 39 % erhöht, während keine statistisch signifikante Änderung der Cmax beobachtet wurde AUC bzw. Cmax. Die klinische Relevanz dieses Anstiegs ist nicht erwiesen. Bei der Anwendung von Ciprofloxacin oder starken CYP1A2-Inhibitoren (z. B. Fluvoxamin) in Kombination mit Erlotinib ist Vorsicht geboten kann reduziert werden.
Die Vorbehandlung oder gleichzeitige Anwendung von Tarceva veränderte die Clearance von CYP3A4-Prototypsubstraten wie Midazolam und Erythromycin nicht, schien jedoch die orale Bioverfügbarkeit von Midazolam um bis zu 24 % zu reduzieren. In einer anderen klinischen Studie veränderte Erlotinib die Pharmakokinetik des gleichzeitig verabreichten Paclitaxel-Substrats von CYP3A4 / 2C8 nicht. Daher sind signifikante Wechselwirkungen mit der Clearance anderer CYP3A4-Substrate unwahrscheinlich.
Die Hemmung der Glucuronidierung kann Wechselwirkungen mit Arzneimitteln verursachen, die Substrate von UGT1A1 sind und ausschließlich auf diesem Weg ausgeschieden werden. Patienten mit reduzierter UGT1A1-Expression oder mit genetischen Veränderungen der Glucuronidierung (z. B. Morbus Gilbert) können erhöhte Serumbilirubinkonzentrationen aufweisen und sollten mit Vorsicht behandelt werden.
Beim Menschen wird Erlotinib in der Leber durch hepatische Cytochrome metabolisiert, hauptsächlich durch CYP3A4 und in geringerem Maße durch CYP1A2 metabolische Clearance von Erlotinib. Es gibt mögliche Wechselwirkungen mit den Wirkstoffen, die von diesen Enzymen metabolisiert werden oder auf diese als Inhibitoren oder Induktoren wirken.
Starke Inhibitoren der CYP3A4-Aktivität reduzieren den Metabolismus von Erlotinib und erhöhen die Plasmakonzentrationen von Erlotinib.In einer klinischen Studie führte die gleichzeitige Anwendung von Erlotinib und Ketoconazol (200 mg zweimal täglich oral über 5 Tage), einem starken CYP3A4-Inhibitor, zu einem Anstieg der Erlotinib-Exposition (86 % der AUC und 69 % der Cmax). Daher ist bei der Anwendung von Erlotinib in Kombination mit einem starken CYP3A4-Inhibitor, wie Azol-Antimykotika (z. B. Ketoconazol, Itraconazol, Voriconazol), Protease-Inhibitoren, Erythromycin oder Clarithromycin, Vorsicht geboten.
Starke Induktoren der CYP3A4-Aktivität erhöhen den Metabolismus von Erlotinib und senken die Plasmakonzentrationen von Erlotinib signifikant.In einer klinischen Studie führte die gleichzeitige Anwendung von Erlotinib und Rifampicin (600 mg / Tag oral über 7 Tage), einem starken Induktor von CYP3A4, zu eine Verringerung der medianen Erlotinib-AUC um 69 % Die gleichzeitige Anwendung von Rifampicin und einer Einzeldosis von 450 mg Tarceva führte zu einer durchschnittlichen Erlotinib-Exposition (AUC) von 57,5 % im Vergleich zu der, die nach Verabreichung einer Einzeldosis von 150 mg Tarceva erreichbar war das Fehlen einer Behandlung mit Rifampicin. Die gleichzeitige Anwendung von Tarceva mit Induktoren von CYP3A4 sollte daher vermieden werden. Bei Patienten, die eine gleichzeitige Behandlung mit Tarceva und einem starken CYP3A4-Induktor wie Rifampicin benötigen, sollte eine Dosiserhöhung auf 300 mg in Betracht gezogen werden, während ihre Sicherheit (einschließlich Nieren- und Leberfunktion sowie Serumelektrolyte) engmaschig überwacht und bei guter Verträglichkeit länger als 2 Wochen kann eine weitere Dosiserhöhung auf 450 mg unter engmaschiger Sicherheitsüberwachung erwogen werden. Eine Expositionsreduktion kann auch bei anderen Induktoren wie Phenytoin, Carbamazepin, Barbituraten oder Johanniskraut auftreten (Hypericum perforatum). Vorsicht ist geboten, wenn diese Wirkstoffe mit Erlotinib kombiniert werden. Wann immer möglich, sollten alternative Behandlungen ohne starke induktive CYP3A4-Aktivität in Betracht gezogen werden.
Antikoagulanzien aus Erlotinib und Cumarin
Bei Patienten, die Tarceva erhielten, wurden Fälle von Wechselwirkungen mit auf Cumarin basierenden Antikoagulanzien, einschließlich Warfarin, die zu einer erhöhten INR (International Normalized Ratio) und Blutungen, die in einigen Fällen tödlich verliefen, führten, berichtet jegliche Veränderungen der Prothrombinzeit oder INR.
Erlotinib und Statine
Die Kombination von Tarceva und einem Statin kann das Risiko einer Statin-induzierten Myopathie, einschließlich einer selten beobachteten Rhabdomyolyse, erhöhen.
Erlotinib und Raucher
Die Ergebnisse einer pharmakokinetischen Wechselwirkungsstudie zeigten eine signifikante Reduktion von 2,8 nach der Verabreichung von Tarceva; Das 1,5- bzw. 9-Fache der AUCinf, Cmax bzw. 24-Stunden-Plasmakonzentration bei Rauchern im Vergleich zu Nichtrauchern (siehe Abschnitt 5.2). Beginn der Behandlung mit Tarceva, andernfalls sind die Plasmakonzentrationen von Erlotinib verringert. Die klinische Wirkung einer reduzierten Exposition ist nicht endgültig belegt, kann aber klinisch bedeutsam sein.
Erlotinib- und P-Glykoprotein-Hemmer
Erlotinib ist ein Substrat von P-Glykoprotein, dem Träger des Wirkstoffs. Die gleichzeitige Anwendung von P-Glykoprotein-Hemmern, beispielsweise Ciclosporin und Verapamil, kann zu einer veränderten Verteilung und/oder Elimination von Erlotinib führen. Die Folgen dieser Wechselwirkung für z. B. ZNS-Toxizität sind nicht bekannt. In solchen Situationen sollte man mit Vorsicht vorgehen.
Erlotinib und Arzneimittel, die den pH-Wert verändern
Erlotinib ist durch eine Abnahme der Löslichkeit bei pH-Werten über 5 gekennzeichnet. Arzneimittel, die den pH-Wert des oberen Gastrointestinaltrakts (GI) verändern, können die Löslichkeit von Erlotinib und folglich seine Bioverfügbarkeit verändern. Die gleichzeitige Anwendung von Erlotinib mit Omeprazol, einem Protonenpumpenhemmer (PPI), verringerte die Erlotinib-Exposition [AUC] und die Spitzenkonzentration [Cmax] um 46 % bzw. 61 % Es wurde keine Veränderung von Tmax oder Halbwertszeit festgestellt. Die gleichzeitige Anwendung von Tarceva mit 300 mg Ranitidin, einem H2-Rezeptor-Antagonisten, verringerte die Erlotinib-Exposition [AUC] und die Spitzenkonzentrationen [Cmax] um 33 % bzw. 54 %. Eine Erhöhung der Tarceva-Dosis bei gleichzeitiger Anwendung mit diesen Arzneimitteln kann dies nicht kompensieren Wenn Tarceva jedoch zeitlich versetzt verabreicht wurde, 2 Stunden vor oder 10 Stunden nach 150 mg Ranitidin 2-mal täglich, sanken die Erlotinib-Exposition [AUC] und die Spitzenkonzentrationen [Cmax] nur um 15 % bzw. 17 % . Die Wirkung von Antazida auf die Resorption von Erlotinib wurde nicht untersucht, aber die Resorption kann beeinträchtigt sein, was zu niedrigeren Plasmaspiegeln führen kann.Zusammenfassend sollte die Kombination von Erlotinib mit Protonenpumpenhemmern vermieden werden. Wenn Antazida während der Behandlung mit Tarceva als notwendig erachtet werden, sollten sie mindestens 4 Stunden vor oder 2 Stunden nach der Tagesdosis von Tarceva eingenommen werden.Wenn Ranitidin in Betracht gezogen wird, sollten sie die beiden Arzneimittel zeitlich versetzt eingenommen werden: Tarceva sollte bei mindestens 2 Stunden vor oder 10 Stunden nach der Verabreichung von Ranitidin.
Erlotinib und Gemcitabin
In einer Phase-Ib-Studie wurden weder signifikante Auswirkungen von Gemcitabin auf die Pharmakokinetik von Erlotinib noch signifikante Auswirkungen von Erlotinib auf die Pharmakokinetik von Gemcitabin beobachtet.
Erlotinib und Carboplatin / Paclitaxel
Erlotinib erhöht die Platinkonzentration. In einer klinischen Studie führte die gleichzeitige Anwendung von Erlotinib mit Carboplatin und Paclitaxel zu einem Anstieg der Gesamtplatin-AUC0-48 um 10,6 %. Obwohl dieser Anstieg statistisch signifikant ist, wird das Ausmaß dieses Unterschieds nicht als klinisch relevant erachtet. In der klinischen Praxis können andere Co-Faktoren auftreten, die zu einer erhöhten Carboplatin-Exposition führen, wie zum Beispiel Nierenversagen. Es wurden keine Auswirkungen von Carboplatin oder . beobachtet Paclitaxel zur Pharmakokinetik von Erlotinib.
Erlotinib und Capecitabin
Capecitabin kann die Erlotinib-Konzentration erhöhen.Bei gleichzeitiger Anwendung von Erlotinib mit Capecitabin kam es zu einem statistisch signifikanten Anstieg der AUC von Erlotinib und einem geringfügigen Anstieg der Cmax im Vergleich zu den Werten, die in einer anderen Studie beobachtet wurden, in der Erlotinib allein verabreicht wurde.Es gibt keine signifikanten Auswirkungen von Erlotinib auf die Pharmakokinetik von Capecitabin.
Erlotinib und Proteasom-Inhibitoren
In Bezug auf den Wirkmechanismus können Proteasom-Inhibitoren einschließlich Bortezomib die Aktivität von EGFR-Inhibitoren einschließlich Erlotinib beeinflussen. Dieser Einfluss wird durch eine begrenzte Verfügbarkeit von klinischen und präklinischen Daten gestützt, die den Abbau von EGFR über das Proteasom hervorheben.
04.6 Schwangerschaft und Stillzeit
Schwangerschaft
Es liegen keine ausreichenden Daten zur Anwendung von Erlotinib bei schwangeren Frauen vor. Tierexperimentelle Studien haben keine Teratogenität oder abnormale Geburten gezeigt. Eine negative Auswirkung auf die Schwangerschaft kann jedoch nicht ausgeschlossen werden, da Studien an Ratten und Kaninchen eine erhöhte embryonale/fetale Letalität gezeigt haben (siehe Abschnitt 5.3) Das potenzielle Risiko für den Menschen ist nicht bekannt.
Frauen im gebärfähigen Alter
Frauen im gebärfähigen Alter sollte geraten werden, während der Behandlung mit Tarceva eine Schwangerschaft zu vermeiden. Während der Behandlung und für mindestens 2 Wochen nach Behandlungsende sollten geeignete Verhütungsmethoden angewendet werden. Bei schwangeren Frauen sollte die Behandlung nur dann fortgesetzt werden, wenn der potenzielle Nutzen für die Mutter das Risiko für den Fötus überwiegt.
Fütterungszeit
Es ist nicht bekannt, ob Erlotinib in die Muttermilch übergeht. Aufgrund der möglichen Schädigung des Neugeborenen sollten Mütter darauf hingewiesen werden, während der Behandlung mit Tarceva nicht zu stillen.
Fruchtbarkeit
Tierexperimentelle Studien haben keine Beeinträchtigung der Fertilität gezeigt. Eine negative Auswirkung auf die Fertilität kann jedoch nicht ausgeschlossen werden, da Tierstudien Auswirkungen auf die Reproduktionsparameter gezeigt haben (siehe Abschnitt 5.3). Das potenzielle Risiko für den Menschen ist unbekannt.
04.7 Auswirkungen auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen
Es wurden keine Studien zur Verkehrstüchtigkeit und zum Bedienen von Maschinen durchgeführt, jedoch wird Erlotinib nicht mit einer Beeinträchtigung der geistigen Fähigkeiten in Verbindung gebracht.
04.8 Nebenwirkungen
Nicht-kleinzelliger Lungenkrebs (Tarceva allein gegeben):
In einer randomisierten Doppelblindstudie (BR.21; Tarceva als Zweitlinientherapie verabreicht) waren die am häufigsten berichteten Nebenwirkungen Hautausschlag (75 %) und Durchfall (54 %), in den meisten Fällen mit einer Intensität von Grad 1 /2 und ohne Zutun handhabbar. Hautausschlag und Diarrhö Grad 3/4 traten bei 9 % bzw. 6 % der mit Tarceva behandelten Patienten auf, beide führten bei 1 % der Patienten zum Abbruch der Studie, Dosierung bei 6 % bzw. 1 % der Patienten. In der Studie BR.21 betrug die mediane Zeit bis zum Auftreten des Hautausschlags 8 Tage und die mediane Zeit bis zum Auftreten des Durchfalls 12 Tage.
Im Allgemeinen äußert sich der Ausschlag als „leichter bis mittelschwerer erythematöser und papulöser-pustulöser Ausschlag, der in sonnenexponierten Bereichen auftreten oder sich verschlimmern kann. Für Patienten, die sich der Sonne aussetzen, kann es ratsam sein, Schutzkleidung zu tragen.“ und/oder Sonnenschutzmittel (zB auf Basis von Mineralstoffen).
Tabelle 1 fasst nach NCI-CTC-Grad (National Cancer Institute Common Toxicity Criteria) die Nebenwirkungen zusammen, die bei Patienten, die mit Tarceva behandelt wurden, im Vergleich zur Placebogruppe in der Zulassungsstudie BR.21 und in mindestens 10 % der Patienten in der Tarceva-Gruppe.
Die folgenden Begriffe werden verwendet, um Nebenwirkungen nach Häufigkeit zu klassifizieren: sehr häufig (≥1 / 10); gewöhnlich (≥1 / 100,
Innerhalb jeder Häufigkeitsgruppe werden die Nebenwirkungen nach abnehmendem Schweregrad aufgeführt.
Tabelle 1: Sehr häufige Nebenwirkungen in Studie BR.21
* Schwere Infektionen mit oder ohne Neutropenie, einschließlich Lungenentzündung, Sepsis und Zellulitis.
** Kann zu Dehydration, Hypokaliämie und Nierenversagen führen.
*** Der Ausschlag umfasste Fälle von akneiformer Dermatitis.
In 2 anderen randomisierten, doppelblinden, placebokontrollierten Phase-III-Studien BO18192 (SATURN) und BO25460 (IUNO); Tarceva wurde als Erhaltungstherapie nach einer Erstlinien-Chemotherapie verabreicht. Diese Studien wurden an insgesamt 1532 Patienten mit fortgeschrittenem, rezidiviertem oder metastasiertem NSCLC nach standardmäßiger Erstlinien-Chemotherapie auf Platinbasis durchgeführt. Es wurden keine neuen Sicherheitsberichte identifiziert.
Die am häufigsten beobachteten Nebenwirkungen bei mit Tarceva behandelten Patienten in den Studien BO18192 und BO25460 waren Hautausschlag und Durchfall (siehe Tabelle 2). In keiner der Studien wurde ein Hautausschlag oder Durchfall vom Grad 4 beobachtet. Hautausschlag und Durchfall führten bei 1 % bzw. 1 % zum Absetzen von Tarceva
Tabelle 2: Häufigste Nebenwirkungen in den Studien BO18192 (SATURN) und BO25460 (IUNO)
* Sicherheitsbewertungsgruppe
In einer offenen Phase-III-Studie, ML 20650 bei 154 Patienten, wurde die Sicherheit von Tarceva als Erstlinientherapie von Patienten mit NSCLC und EGFR-aktivierenden Mutationen bei 75 Patienten untersucht; es wurden keine neuen diesbezüglichen Berichte identifiziert.
Die am häufigsten beobachteten Nebenwirkungen bei mit Tarceva behandelten Patienten mit ML 20650 waren Hautausschlag und Durchfall (jeden Grades, 80 % bzw. 57 %), meist Intensitätsgrade 1/2 und beherrschbar % der Patienten Es wurde kein Fall von Hautausschlag oder Durchfall vom Grad 4 beobachtet Hautausschlag und Durchfall führten beide dazu, dass Tarceva bei 1 % der Patienten abgesetzt wurde. Dosisanpassungen (Unterbrechungen oder Reduzierungen) aufgrund von Hautausschlag und Durchfall waren bei 11 % bzw. 7 % der Patienten erforderlich.
Bauchspeicheldrüsenkrebs (Tarceva zusammen mit Gemcitabin verabreicht):
Die häufigsten Nebenwirkungen in der Zulassungsstudie PA.3 bei Patienten mit Bauchspeicheldrüsenkrebs, die mit Tarceva 100 mg und Gemcitabin behandelt wurden, waren Müdigkeit, Hautausschlag und Durchfall. Im Behandlungsarm mit Tarceva plus Gemcitabin wurden jeweils bei 5 % der Patienten Hautausschlag und Durchfall Grad 3/4 berichtet.Die mediane Zeit bis zum Auftreten von Hautausschlag und Durchfall betrug 10 bzw. 15 Tage. Hautausschlag und Durchfall führten jeweils bei 2 % der Patienten zu einer Dosisreduktion und bei bis zu 1 % der Patienten, die Tarceva plus Gemcitabin erhielten, zum Abbruch der Studie.
In der Zulassungsstudie PA.3 traten Nebenwirkungen häufiger (≥ 3 %) bei Patienten auf, die mit Tarceva 100 mg plus Gemcitabin behandelt wurden als in der Placebo- plus Gemcitabin-Gruppe und bei mindestens 10 % der Patienten in der Tarceva-Gruppe 100 mg plus Gemcitabin sind in Tabelle 3 zusammengefasst, basierend auf den Common Toxicity Criteria (NCI-CTC) des National Cancer Institute.
Die folgenden Begriffe werden verwendet, um Nebenwirkungen nach Häufigkeit zu klassifizieren: sehr häufig (≥1 / 10); gewöhnlich (≥1 / 100,
Innerhalb jeder Häufigkeitsgruppe werden die Nebenwirkungen nach abnehmendem Schweregrad aufgeführt.
Tabelle 3: Sehr häufige Nebenwirkungen in Studie PA.3 (100-mg-Kohorte)
* Schwere Infektionen mit oder ohne Neutropenie, einschließlich Lungenentzündung, Sepsis und Zellulitis.
** Kann zu Dehydration, Hypokaliämie und Nierenversagen führen.
*** Der Ausschlag umfasste Fälle von akneiformer Dermatitis.
Sonstige Anmerkungen:
Die Sicherheitsbewertung von Tarceva basiert auf Daten von mehr als 1500 Patienten, die mit mindestens einer Dosis von 150 mg Tarceva allein behandelt wurden, und von mehr als 300 Patienten, die mit Tarceva 100 oder 150 mg in Kombination mit Gemcitabin behandelt wurden.
Die folgenden Nebenwirkungen wurden bei Patienten beobachtet, die mit Tarceva als Monotherapie behandelt wurden, und bei Patienten, die gleichzeitig mit Tarceva mit einer Chemotherapie behandelt wurden.
Sehr häufige Nebenwirkungen aus den Studien BR 21 und PA 3 werden in den Tabellen 1 und 3 berichtet, andere Nebenwirkungen, einschließlich derjenigen aus anderen Studien, sind in Tabelle 4 zusammengefasst.
Innerhalb jeder Häufigkeitsgruppe werden die Nebenwirkungen nach abnehmendem Schweregrad aufgeführt.
Tabelle 4: Nebenwirkungen nach Häufigkeitskategorie
1 In Studie PA.3.
2 Einschließlich Wimpern, die nach innen wachsen, übermäßiges Wachstum und Verdickung der Wimpern.
3 Einschließlich tödlicher Fälle bei Patienten, die Tarceva zur Behandlung von NSCLC oder anderen fortgeschrittenen soliden Tumoren einnehmen (siehe Abschnitt 4.4). Eine höhere Inzidenz wurde bei Patienten japanischer Abstammung beobachtet.
4 In klinischen Studien wurden einige Fälle mit der gleichzeitigen Gabe von Warfarin (siehe Abschnitt 4.5) und manchmal mit der gleichzeitigen Gabe von NSAR in Verbindung gebracht.
5 Einschließlich Anstieg der Alanin-Aminotransferase (ALT), Aspartat-Aminotransferase (AST) und Bilirubin). Diese waren meist leichter oder mittelschwerer Natur, vorübergehender Natur oder mit Lebermetastasen verbunden.
6 Einschließlich tödlicher Fälle. Eine vorbestehende Lebererkrankung oder die gleichzeitige Anwendung von hepatotoxischen Arzneimitteln wurden als Störfaktoren angesehen (siehe Abschnitt 4.4).
7 Einschließlich tödlicher Fälle (siehe Abschnitt 4.4).
Meldung von vermuteten Nebenwirkungen
Die Meldung vermuteter Nebenwirkungen, die nach der Zulassung des Arzneimittels aufgetreten sind, ist wichtig, da sie eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels ermöglicht. Angehörige von Gesundheitsberufen werden gebeten, jeden Verdachtsfall einer Nebenwirkung über das nationale Meldesystem zu melden. "Adresse https: //www.aifa.gov.it/content/segnalazioni-reazioni-avverse.
04.9 Überdosierung
Symptome
Orale Einzeldosen von Tarceva bis zu 1000 mg Erlotinib bei gesunden Probanden und bis zu 1600 mg bei Krebspatienten wurden vertragen. Wiederholte Dosen von 200 mg zweimal täglich wurden bei gesunden Probanden nach einigen Tagen der Verabreichung schlecht vertragen. Basierend auf den Daten aus diesen Studien ist es möglich, dass bei höheren als den empfohlenen Dosen schwerwiegende Nebenwirkungen wie Durchfall, Hautausschlag und möglicherweise erhöhte hepatische Aminotransferase-Aktivität auftreten können.
Verwaltung
Bei Verdacht auf eine Überdosierung sollte die Anwendung von Tarceva abgebrochen und eine symptomatische Behandlung eingeleitet werden.
05.0 PHARMAKOLOGISCHE EIGENSCHAFTEN
05.1 Pharmakodynamische Eigenschaften
Pharmakotherapeutische Gruppe: Proteinkinase-Inhibitor, antineoplastisches Arzneimittel.
ATC-Code: L01XE03.
Wirkmechanismus
Erlotinib ist ein epidermaler Wachstumsfaktorrezeptor / humaner epidermaler Wachstumsfaktorrezeptor Typ I (EGFR, auch bekannt als HER1)-Tyrosinkinase-Hemmer. Erlotinib ist ein potenter Inhibitor der intrazellulären Phosphorylierung von EGFR. EGFR wird auf der Zelloberfläche von normalen und Tumorzellen exprimiert.In nicht-klinischen Modellen führt die Hemmung von EGFRs Phosphotyrosin zu Zellstaseund/oder zum Tod.
EGFR-Mutationen können zu einer „konstitutiven Aktivierung der anti-apoptotischen und proliferativen Signalwege führen. Die starke Wirksamkeit von Erlotinib bei der Blockierung der EGFR-vermittelten Signaltransduktion bei diesen EGFR-Mutations-positiven Tumoren wird auf die enge Verbindung zwischen Erlotinib an der ATP-Bindungsstelle zurückgeführt in der mutierten Kinasedomäne von EGFR.Aufgrund der Blockade des Downstream-Transduktionssignals wird die Zellproliferation gestoppt und der Zelltod wird durch den intrinsischen apoptotischen Weg induziert. In Mausmodellen mit deutlicher Expression dieser aktivierenden EGFR-Mutationen wird eine Tumorregression beobachtet.
Klinische Wirksamkeit
• Erstlinientherapie von nicht-kleinzelligem Lungenkrebs (NSCLC) bei Patienten mit aktivierenden EGFR-Mutationen (Tarceva allein verabreicht):
Die Wirksamkeit von Tarceva bei Erstlinienpatienten mit aktivierenden EGFR-Mutationen bei NSCLC wurde in einer randomisierten, offenen Phase-III-Studie (ML20650, EURTAC) nachgewiesen. Diese Studie wurde an kaukasischen Patienten mit metastasiertem oder lokal fortgeschrittenem NSCLC (Stadium IIIB und IV) durchgeführt, die zuvor keine Chemotherapie oder systemische antineoplastische Therapie gegen ihre Erkrankung erhalten hatten und Mutationen in der Tyrosinkinase-Domäne von EGFR (Exon-Deletion) aufwiesen oder Exon 21-Mutation) Die Patienten wurden 1:1 randomisiert und erhielten Tarceva 150 mg täglich oder bis zu 4 Zyklen einer platinbasierten Korsett-Chemotherapie.
Der primäre Endpunkt war das vom Prüfarzt beurteilte PFS.
Die Wirksamkeitsergebnisse sind in Tabelle 5 zusammengefasst.
Tabelle 5: Wirksamkeitsergebnisse von Tarceva im Vergleich zur Chemotherapie in der Studie ML20650 (EURTAC)
CR = vollständige Antwort; RP = Teilantwort.
* Es wurde eine 58 %ige Verringerung des Risikos für Krankheitsprogression oder Tod beobachtet.
** Die Gesamtübereinstimmungsrate zwischen der Beurteilung des PFS durch den Prüfarzt und das IRC betrug 70 %.
*** Eine „hohe Cross-over-Rate“ wurde bei 82 % der Patienten im Chemotherapie-Arm beobachtet, die eine Folgetherapie mit einem EGFR-assoziierten Tyrosinkinase-Inhibitor erhielten, und alle bis auf 2 Patienten wurden mit Tarceva behandelt.
- Erhaltungstherapie von NSCLC nach Erstlinien-Chemotherapie (Tarceva wird als Monotherapie verabreicht):
Die Wirksamkeit und Sicherheit von Tarceva als Erhaltungstherapie nach einer Erstlinien-Chemotherapie bei NSCLC wurden in einer randomisierten, doppelblinden, placebokontrollierten Studie (BO18192, SATURN) untersucht, an der 889 Patienten mit lokal fortgeschrittenem NSCLC teilnahmen, die nach 4 Zyklen einer Chemotherapie auf Platinbasis mit zwei Wirkstoffen. Die Patienten wurden 1:1 einer Behandlung mit Tarceva 150 mg oder Placebo oral einmal täglich bis zur Progression zugeteilt. L "Der Hauptendpunkt der Studie war das progressionsfreie Überleben (PFS) in allen" Patienten. Die demografischen und Krankheitsmerkmale bei Aufnahme waren zwischen den beiden Behandlungsarmen ausgewogen Patienten mit ECOG PS > 1, signifikanten hepatischen oder renalen Komorbiditäten wurden nicht in die Studie eingeschlossen.
In dieser Studie zeigte die gesamte Population einen Vorteil für den primären Endpunkt PFS (HR = 0,71 p .).
67 % der mit Placebo behandelten Patienten in der EGFR-Mutations-positiven Untergruppe erhielten EGFR-TKI-Inhibitoren in der zweiten oder nachfolgenden Behandlungslinie.
Studie BO25460 (IUNO) wurde an 643 Patienten mit fortgeschrittenem NSCLC ohne aktivierende EGFR-Mutationen (Exon 19-Deletion oder Exon 21 L858R-Mutation) durchgeführt, die nach vier Zyklen platinbasierter Chemotherapie keine Krankheitsprogression gezeigt hatten.
Das Ziel der Studie war es, das Gesamtüberleben von Erlotinib als Erstlinientherapie mit Erlotinib zum Zeitpunkt der Krankheitsprogression zu vergleichen Die Studie erreichte den primären Endpunkt nicht. Das OS von Tarceva bei der Erhaltungstherapie war dem von Tarceva als Zweitlinientherapie bei Patienten, deren Tumor keine aktivierende EGFR-Mutation aufwies, nicht überlegen (HR = 1,02, 95 %-KI, 0,85-1, 22, p = 0,82) Der sekundäre Endpunkt des progressionsfreien Überlebens (PFS) unterschied sich bei der Erhaltungstherapie nicht zwischen Tarceva und Placebo (HR = 0,94, 95 %-KI, 0,80-1,11; p = 0,48).
Basierend auf den Daten aus der Studie BO25460 (IUNO) wird die Anwendung von Tarceva als Erstlinien-Erhaltungstherapie bei Patienten ohne aktivierende EGFR-Mutation nicht empfohlen.
- Behandlung von NSCLC nach Versagen mindestens einer vorherigen Chemotherapielinie (Tarceva allein verabreicht):
Die Wirksamkeit und Sicherheit von Tarceva als Zweit-/Drittlinientherapie wurde in einer randomisierten, doppelblinden, placebokontrollierten Studie (BR.21) bei 731 Patienten mit lokal fortgeschrittenem oder metastasiertem NSCLC nach Versagen mindestens einer Chemotherapie nachgewiesen wurden randomisiert 2:1 einer Behandlung mit Tarceva 150 mg oder Placebo oral einmal täglich zugeteilt.Zu den Studienendpunkten gehörten das Gesamtüberleben, das progressionsfreie Überleben (PFS), das Ansprechen, die Dauer des Ansprechens, die Zeit bis zur Verschlechterung der lungenkrebsbedingten Symptome (Husten, Dyspnoe) und Schmerzen) und Sicherheit.Der primäre Endpunkt war das Überleben.
Die demografischen Merkmale waren zwischen den beiden Behandlungsgruppen gut ausgewogen. Etwa zwei Drittel der Patienten waren männlich, etwa ein Drittel hatte einen ECOG-Performance-Status (PS) bei Aufnahme von 2 bzw. 9 % und einen ECOG-PS bei Aufnahme von 3. 93 % bzw. 92 % aller Patienten Die Tarceva-Gruppe und die Placebo-Gruppe waren zuvor mit einem platinbasierten Regime behandelt worden und 36 % bzw. 37 % aller Patienten waren zuvor mit Taxanen behandelt worden.
Die korrigierte Hazard Ratio (HR) für Todesfälle in der Tarceva-Gruppe gegenüber der Placebo-Gruppe betrug 0,73 (95%-KI: 0,60–0,87) (p = 0,001). 31,2 % bzw. 21,5% der Patienten in der Tarceva- bzw. der Placebo-Gruppe waren nach 12 Monaten noch am Leben. Das mediane Gesamtüberleben betrug 6,7 Monate in der Tarceva-Gruppe (95%-KI: 5,5–7,8 Monate) im Vergleich zu 4,7 Monaten in der Placebo-Gruppe (95%-KI: 4,1–6, 3 Monate).
Die Wirkung auf das Gesamtüberleben wurde in verschiedenen Patientenuntergruppen untersucht.Die Wirkung von Tarceva auf das Gesamtüberleben war bei Patienten mit einem Ausgangsleistungsstatus (ECOG) von 2-3 (HR = 0,77, KI 95 % 0,6-1,0) oder 0- ähnlich. 1 (HR = 0,73, 95 %-KI 0,6–0,9), bei männlichen Patienten (HR = 0,76, 95 %-KI 0, 6–0,9) oder weiblich (HR = 0,80, 95 %-KI 0,6–1,1), bei Patienten unter 65 Jahre alt (HR = 0,75, 95 %-KI 0,6–0,9) oder bei älteren Patienten (HR = 0,79, 95 %-KI 0,6–1,0), bei Patienten, die mit einem früheren Regime behandelt wurden (HR = 0,76, 95 %-KI % 0,6 -1.0) oder mit mehr als einer vorherigen Therapie (HR = 0,75, 95 %-KI 0,6-1,0), bei kaukasischen Patienten (HR = 0,79, 95 %-KI 0,6-1,0) oder Asiaten (HR = 0,61, 95 %-KI 0,4- 1,0), bei Patienten mit Adenokarzinom (HR = 0,71, 95 % CI 0,6–0, 9) oder Plattenepithelkarzinom (HR = 0,67, 95 % CI 0,5–0,9), jedoch nicht bei Patienten mit anderer Histologie (HR 1,04, 95 % KI 0,7-1,5 ), bei Patienten mit Erkrankung im Stadium IV bei Diagnose (HR = 0,92, 95 %-KI 0,7-1,2) oder Stadium
Bei 45 % der Patienten mit bekanntem EGFR-Expressionsstatus betrug die Hazard Ratio 0,68 (95 % KI 0,49 – 0,94) für Patienten mit EGFR-positiven Tumoren und 0, 93 (95 % KI 0,63 – 1,36) für Patienten mit EGFR-negativen Tumoren (von IHC unter Verwendung des EGFR pharmDx-Kits als EGFR-negativ diejenigen mit weniger als 10 % Tumorzellmarkierung definiert). Bei den verbleibenden 55 % der Patienten mit unbekanntem EGFR-Expressionsstatus betrug die Hazard Ratio 0,77 (95 % CI 0,61-0,98).
Das mediane PFS betrug 9,7 Wochen in der Tarceva-Gruppe (95%-KI 8,4-12,4 Wochen) im Vergleich zu 8,0 Wochen in der Placebo-Gruppe (95%-KI 7,9-8,1 Wochen).
In der Tarceva-Gruppe betrug die objektive Ansprechrate nach RECIST 8,9 % (95 %-KI 6,4-12,0) Die ersten 330 Patienten wurden zentral beurteilt (Antwortrate 6, 2 %) und 401 Patienten wurden vom Prüfarzt beurteilt (Ansprechrate 11,2 % ).
Die mediane Ansprechdauer betrug 34,3 Wochen, mit einem Minimum von 9,7 und einem Maximum von 57,6+ Wochen. 44,0 % der Patienten erreichten in der Tarceva-Gruppe ein vollständiges, teilweises Ansprechen oder eine Stabilisierung der Krankheit, verglichen mit 27,5 % der Patienten in der Placebo-Gruppe (p = 0,004).
Ein Überlebensvorteil mit Tarceva wurde auch bei Patienten beobachtet, die kein objektives Ansprechen des Tumors (RECIST-Kriterien) erreichten. Dies wurde durch eine Hazard Ratio für den Tod von 0,82 (95% CI 0,68-0,99) bei Patienten belegt, die als beste Reaktion eine Stabilisierung oder Progression der Erkrankung erreichten.
Tarceva induzierte symptomatische Vorteile, indem es die Zeit bis zur Verschlechterung von Husten, Atemnot und Schmerzen im Vergleich zu Placebo signifikant verlängerte.
- Bauchspeicheldrüsenkrebs (Tarceva zusammen mit Gemcitabin in Studie PA.3):
Die Wirksamkeit und Sicherheit von Tarceva in Kombination mit Gemcitabin als Erstlinientherapie wurden in einer randomisierten, doppelblinden, placebokontrollierten Studie bei Patienten mit lokal fortgeschrittenem, nicht resezierbarem oder metastasiertem Bauchspeicheldrüsenkrebs untersucht täglich auf kontinuierlicher Basis und iv Gemcitabin (1000 mg / m2, Zyklus 1 - Tage 1, 8, 15, 22, 29, 36 und 43 eines 8-Wochen-Zyklus; 2 und folgende Zyklen - Tage 1, 8 und 15 eines 4-wöchigen Zyklus [zugelassene Dosierung und Zeitplan für Bauchspeicheldrüsenkrebs, siehe Fachinformation Gemcitabin]). Tarceva oder Placebo wurde einmal täglich oral eingenommen, bis die Krankheit fortschreitet oder eine inakzeptable Toxizität aufgetreten ist. Der primäre Endpunkt war das Gesamtüberleben.
Die demografischen und Krankheitsmerkmale der Patienten bei Aufnahme waren für die beiden Behandlungsgruppen Tarceva 100 mg plus Gemcitabin oder Placebo plus Gemcitabin ähnlich, mit Ausnahme eines etwas höheren Anteils von Frauen im Erlotinib-/Gemcitabin-Arm im Vergleich zum Placebo-/Gemcitabin-Arm. :
Das Überleben wurde in der Intent-to-Treat-Population auf der Grundlage von Follow-up-Überlebensdaten bewertet. Die Ergebnisse sind in der folgenden Tabelle beschrieben (die Ergebnisse für die Patientengruppen mit metastasierter und lokal fortgeschrittener Erkrankung stammen aus einer explorativen Subgruppenanalyse).
Patienten mit einem günstigen klinischen Ausgangszustand (niedrige Schmerzintensität, gute Lebensqualität und guter PS) könnten mehr von Tarceva profitieren, wie eine „Post-hoc-Analyse“ zeigt. Der Nutzen ergibt sich hauptsächlich aus dem Vorliegen einer geringen Schmerzintensität .
In einer Post-hoc-Analyse hatten die mit Tarceva behandelten Patienten, die einen Hautausschlag entwickelten, ein längeres Gesamtüberleben im Vergleich zu Patienten, die keinen Hautausschlag entwickelten (medianes OS 7,2 Monate vs. 5 Monate, HR: 0,61).
90 % der Patienten, die Tarceva einnahmen, entwickelten innerhalb der ersten 44 Tage einen Hautausschlag. Die mediane Zeit bis zum Auftreten des Hautausschlags betrug 10 Tage.
Kinder und Jugendliche
Die Europäische Arzneimittel-Agentur hat auf die Verpflichtung zur Vorlage der Ergebnisse von Studien mit Tarceva in allen Untergruppen der pädiatrischen Population für Indikationen bei nicht-kleinzelligem Lungenkrebs und Bauchspeicheldrüsenkrebs verzichtet (siehe Abschnitt 4.2 für Informationen zur „pädiatrischen Anwendung“).
05.2 „Pharmakokinetische Eigenschaften
AbsorptionMaximale Plasmakonzentrationen von Erlotinib werden etwa 4 Stunden nach oraler Verabreichung erreicht.Eine Studie an gesunden Probanden ergab eine geschätzte absolute Bioverfügbarkeit von 59 %. Nahrung kann die Exposition nach oraler Gabe erhöhen.
Verteilung: Erlotinib hat ein mittleres scheinbares Verteilungsvolumen von 232 l und wird im menschlichen Tumorgewebe verteilt. In einer Studie, die an 4 Patienten (3 mit nicht-kleinzelligem Lungenkrebs (NSCLC) und 1 mit Kehlkopfkrebs) durchgeführt wurde, die mit 150 mg Tarceva pro Tag oral behandelt wurden, wurden die Proben, die durch chirurgische Entfernung des Tumors am 9. der Behandlung zeigten Erlotinib-Konzentrationen innerhalb des Tumors von durchschnittlich 1,185 ng/g Gewebe, was insgesamt einem Mittelwert von 63 % (Bereich: 5–161 %) der im Steady State beobachteten maximalen Plasmakonzentration entspricht im Tumor bei Konzentrationen von durchschnittlich 160 ng / g Gewebe, was insgesamt einem Mittelwert von 113% (Bereich: 88-130%) der im Steady State beobachteten Plasma-Spitzenkonzentration entspricht Die Plasmaproteinbindung beträgt etwa 95%. Erlotinib bindet an Serumalbumin und das saure Alpha-1-Glykoprotein (AAG).
Biotransformation: Beim Menschen wird Erlotinib in der Leber durch hepatische Cytochrome metabolisiert, hauptsächlich durch CYP3A4 und in geringerem Maße durch CYP1A2.Die durch CYP3A4 im Darm, CYP1A1 in der Lunge und 1B1 im Tumorgewebe vermittelte extrahepatische Metabolisierung trägt möglicherweise zur metabolischen Clearance von Erlotinib bei.
Drei Hauptstoffwechselwege wurden identifiziert: 1) O-Demethylierung einer oder beider Seitenketten, gefolgt von Oxidation zu Carbonsäuren, 2) Oxidation der acetylenischen Fraktion, gefolgt von Hydrolyse zu Arylcarbonsäure, und 3) aromatische Hydroxylierung des Phenylacetylens Fraktion. Die wichtigsten Erlotinib-Metaboliten OSI-420 und OSI-413, die durch O-Demethylierung einer der Seitenketten produziert werden, zeigten in nicht-klinischen Analysen eine ähnliche Wirksamkeit wie Erlotinib. in vitro und in Tumormodellen in vivo. Sie sind im Plasma in Konzentrationen von weniger als 10 % der von Erlotinib vorhanden und weisen eine ähnliche Pharmakokinetik wie Erlotinib auf.
Beseitigung: Erlotinib wird hauptsächlich über die Fäzes ausgeschieden (> 90 %), während die renale Elimination nur einen kleinen Bruchteil (ca. 9 %) der oral verabreichten Menge ausmacht. Weniger als 2 % der oral verabreichten Dosis werden unverändert ausgeschieden von 591 Patienten, die mit Tarceva als Monotherapie behandelt wurden, zeigt eine mittlere scheinbare Clearance von 4,47 l / h mit einer medianen Halbwertszeit von 36,2 Stunden, daher ist zu erwarten, dass die Plasmakonzentration im Steady-State in etwa 7 oder 8 Tagen erreicht wird.
Pharmakokinetik in speziellen Populationen:
Basierend auf der populationspharmakokinetischen Analyse wurden keine klinisch signifikanten Korrelationen zwischen der scheinbaren erwarteten Clearance und dem Alter, Körpergewicht, Geschlecht oder ethnischer Zugehörigkeit des Patienten beobachtet. Patientenbezogene Faktoren, die eine Korrelation mit der Pharmakokinetik von Erlotinib zeigten, waren Gesamtbilirubin, „AAG und“ als Raucher Erhöhte Serum-Gesamtbilirubin- und AAG-Konzentrationen waren mit einer verringerten Erlotinib-Clearance verbunden. Die klinische Relevanz dieser Unterschiede ist unklar. Allerdings war die Erlotinib-Clearance bei Rauchern erhöht.
Dies wurde durch eine pharmakokinetische Studie an gesunden Nichtrauchern und Zigarettenrauchern bestätigt, die eine orale Einzeldosis von 150 mg Erlotinb erhielten. Der geometrische Mittelwert von Cmax betrug 1056 ng/ml bei Nichtrauchern und 689 ng/ml bei Rauchern mit einem mittleren Verhältnis von Rauchern zu Nichtrauchern von 65,2 % (95%-KI: 44,3 – 95,9; p = 0,031). Der geometrische Mittelwert der AUC0-inf betrug 18726 ng • h / ml bei Nichtrauchern und 6718 ng • h / ml bei Rauchern mit einem mittleren Verhältnis von 35,9 % (95 % KI: 23,7 - 54, 3; p .).
In der zulassungsrelevanten Phase-III-NSCLC-Studie erreichten Raucher eine Steady-State-Plasmakonzentration von Erlotinib von 0,65 µg/ml (n = 16), ungefähr 2-mal niedriger als bei ehemaligen Rauchern oder Patienten, die nie geraucht haben (1,28 µg/ml, n = 108). Dieser Effekt wurde von einer 24 %igen Erhöhung der scheinbaren Plasmaclearance von Erlotinib begleitet. In einer Phase-I-Dosiseskalationsstudie bei rauchenden NSCLC-Patienten zeigten pharmakokinetische Analysen im Steady-State einen dosisproportionalen Anstieg der Erlotinib-Exposition durch Erhöhung der Tarceva-Dosis von 150 mg auf die maximal verträgliche Dosis von 300 mg. Zustand Plasmakonzentration bei der 300-mg-Dosis bei Rauchern betrug 1,22 μg / ml (n = 17).
Aufgrund der Ergebnisse von pharmakokinetischen Studien sollte Rauchern geraten werden, während der Einnahme von Tarceva mit dem Rauchen aufzuhören, da sonst die Plasmakonzentrationen reduziert werden können.
Basierend auf einer populationspharmakokinetischen Analyse scheint das Vorhandensein eines Opioids die Exposition um etwa 11 % zu erhöhen.
Eine zweite pharmakokinetische Populationsanalyse wurde durchgeführt, einschließlich Erlotinib-Daten von 204 Patienten mit Bauchspeicheldrüsenkrebs, die mit Erlotinib und Gemcitabin behandelt wurden.Diese Analyse zeigte, dass Kovarianten, die die Erlotinib-Clearance bei Studienpatienten mit Bauchspeicheldrüsenkrebs beeinflussten, denen bei der vorherigen pharmakokinetischen Analyse der Monotherapie sehr ähnlich waren. Es wurden keine neuen kovariaten Effekte identifiziert. Die gleichzeitige Anwendung von Gemcitabin hatte keinen Einfluss auf die Plasmaclearance von Erlotinib.
Kinder und Jugendliche: Es wurden keine spezifischen Studien an pädiatrischen Patienten durchgeführt
Ältere Patienten: Bei älteren Patienten wurden keine spezifischen Studien durchgeführt.
Leberinsuffizienz: Die Clearance von Erlotinib erfolgt überwiegend über die Leber. Bei Patienten mit soliden Tumoren und mäßiger Leberfunktionsstörung (Child-Pugh-Score von 7-9) betrugen die geometrischen Mittelwerte von AUC0-t und Cmax von Erlotinib 27.000 ng • h / ml und 805 ng / ml, während sie 29300 ng • h / ml und 1090 ng / ml bei Patienten mit adäquater Leberfunktion, einschließlich solcher mit primärem Leberkrebs oder Lebermetastasen Obwohl Cmax bei Patienten mit mäßiger Leberfunktionsinsuffizienz statistisch signifikant niedriger war, wird dieser Unterschied nicht als klinisch relevant angesehen die Auswirkung einer schweren Leberfunktionsstörung auf die Pharmakokinetik von Erlotinib. In der populationspharmakokinetischen Analyse ist ein Anstieg der Gesamtbilirubinkonzentration im Serum mit einer Verlangsamung der Erlotinib-Clearance verbunden.
Nierenversagen: Die renale Ausscheidung von Erlotinib und seinen Metaboliten ist nicht signifikant, da weniger als 9 % einer Einzeldosis mit dem Urin ausgeschieden werden Klinisch signifikante Korrelationen zwischen Erlotinib-Clearance und Kreatinin-Clearance, es liegen jedoch keine Daten für Patienten mit Kreatinin-Clearance vor
05.3 Präklinische Sicherheitsdaten
Zu den nach chronischer Verabreichung bei mindestens einer Tierart oder einer Studie beobachteten Wirkungen gehören solche an der Hornhaut (Atrophie, Ulzeration), an der Haut (follikuläre Degeneration und Entzündung, Rötung und Alopezie), am Eierstock (Atrophie), an der Leber ( Lebernekrose), an der Niere (nierenpapilläre Nekrose und tubuläre Dilatation) und am Magen-Darm-Trakt (verzögerte Magenentleerung und Durchfall). ein Anstieg von ALT, AST und Bilirubin war verbunden, und diese Daten wurden für Expositionen deutlich unterhalb der klinisch relevanten Expositionen erhoben.
Basierend auf dem Wirkmechanismus ist Erlotinib potenziell teratogen. Daten aus Reproduktionstoxizitätsstudien an Ratten und Kaninchen bei Dosen nahe der maximal verträglichen Dosis und/oder maternal toxischen Dosen wiesen auf Reproduktionstoxizität (Embryotoxizität bei der Ratte) hin, embryonale Resorption und Fetotoxizität bei Kaninchen) und Entwicklung (verringertes Wachstum und Überleben von Jungtieren bei Ratten), zeigte jedoch keine Teratogenität oder Beeinträchtigung der Fertilität. Diese Ergebnisse wurden bei klinisch relevanten Expositionen beobachtet.
Konventionelle Genotoxizitätsstudien von Erlotinib sind fehlgeschlagen. 2-Jahres-Karzinogenitätsstudien, die an Ratten und Mäusen mit Erlotinib bis zu Konzentrationen oberhalb der beim Menschen verwendeten therapeutischen Konzentrationen (bis zum 2-fachen bzw. 10-fachen, bezogen auf Cmax und/oder AUC) durchgeführt wurden, waren negativ.
Bei Ratten wurde nach UV-Bestrahlung eine leichte phototoxische Hautreaktion beobachtet.
06.0 PHARMAZEUTISCHE INFORMATIONEN
06.1 Hilfsstoffe
Kern des Tablets:
Lactose-Monohydrat
Mikrokristalline Cellulose (E460)
Natriumstärkeglycolat Typ A
Natriumlaurylsulfat
Magnesiumstearat (E470 b)
Tablettenbeschichtung:
Hydroxypropylcellulose (E463)
Titandioxid (E171)
Macrogol
Hypromellose (E464)
06.2 Inkompatibilität
Nicht relevant.
06.3 Gültigkeitsdauer
4 Jahre.
06.4 Besondere Vorsichtsmaßnahmen für die Aufbewahrung
Für dieses Arzneimittel sind keine besonderen Lagerungsbedingungen erforderlich.
06.5 Art der unmittelbaren Verpackung und Inhalt des Packstücks
Mit Aluminiumfolie versiegelte PVC-Blisterpackung mit 30 Tabletten.
06.6 Gebrauchs- und Handhabungshinweise
Keine besonderen Hinweise zur Entsorgung.
Nicht verwendete Arzneimittel und Abfälle aus diesem Arzneimittel müssen gemäß den örtlichen Vorschriften entsorgt werden.
07.0 INHABER DER MARKETING-ERLAUBNIS
Roche Registration Limited
6 Falkenweg
Shire-Park
Welwyn Gartenstadt
AL7 1TW
Vereinigtes Königreich
08.0 NUMMER DER MARKETING-ERLAUBNIS
EU / 1/05/311/003
036871034
09.0 DATUM DER ERSTEN GENEHMIGUNG ODER ERNEUERUNG DER GENEHMIGUNG
Datum der Erstzulassung: 19. September 2005
Datum der letzten Verlängerung: 19. September 2010
10.0 DATUM DER ÜBERARBEITUNG DES TEXTs
Januar 2016