Was ist das Marfan-Syndrom?
Das Marfan-Syndrom beschreibt eine komplexe erbliche Erkrankung des Bindegewebes, die vor allem die Augen, das Herz-Kreislauf-System und den Bewegungsapparat betrifft. Da jedoch jedes Organ aus Bindegewebe besteht, kann das Marfan-Syndrom idealerweise das Wachstum und die Funktion jeder anatomischen Stelle zerstören und stark beeinträchtigen.
Das Syndrom wird autosomal-dominant vererbt: Wir haben es also mit einer schwerwiegenden genetischen Erkrankung zu tun, die einen extrem variablen "phänotypischen Ausdruck" hat (die Defekte können von Familie zu Familie oder von Patient zu Patient enorm unterschiedlich sein).
Was das Marfan-Syndrom auslöst, ist die Veränderung des FBN1-Gens (auf Chromosom 15), das für Fibrillin-1 kodiert, ein sehr wichtiges Bindeglykoprotein, das den strukturellen Träger für Mikrofibrillen darstellt.
Mikrofibrillen: Mikrofibrillen bestehen aus Fibrillin und sind in der extrazellulären Matrix vorhanden, in der sie ein Geflecht für die Einlagerung von Elastin in die elastischen Fasern bilden. Obwohl im Körper allgegenwärtig, sind Mikrofibrillen vor allem in der Aorta, den Bändern und Zonula der Ziliarkörper (auf Augenebene).
Da es sich um eine autosomal-dominant vererbte Erkrankung handelt, sind nur Kinder vom Marfan-Syndrom betroffen, die von beiden Elternteilen ein verändertes FBN-1-Gen geerbt haben. Dennoch ist die Krankheit in einem von vier Fällen das Ergebnis spontaner Mutationen bei Patienten, die keine Familienanamnese haben.
Der Name der Krankheit leitet sich von dem französischen Kinderarzt ab, der sie 1896 erstmals beschrieb (A. Marfan). Danach musste bis 1991 gewartet werden, um das veränderte Gen zu identifizieren, das an der symptomatologischen Manifestation beteiligt war: Der Entdecker war F. Ramirez.
Schau das Video
- Schau dir das Video auf youtube an
Ursachen
Wir haben erwähnt, dass das Marfan-Syndrom der unmittelbare Ausdruck der Mutation eines Gens ist, das für Fibrillin-1 kodiert.
FIBRILLIN 1 ist eine Glykoproteinkomponente von Elastin, die für die Gewährleistung und Aufrechterhaltung der Gewebeelastizität und -festigkeit unerlässlich ist.Unter physiologischen Bedingungen bindet Fibrillin 1 an ein anderes Protein, bekannt als TGF-beta (oder Transforming Growth Factor Beta). TGF-beta scheint an schädlichen Prozessen beteiligt zu sein, die die glatte Gefäßmuskulatur und die extrazelluläre Matrix betreffen. Ausgehend von diesen Annahmen sind einige Autoren überzeugt, dass das Marfan-Syndrom neben der Mutation des FBN-1-Gens auch auf einen Überschuss an TGF-beta insbesondere in Aorta, Herzklappen und Lunge zurückzuführen ist.

Vorfall
Es wird geschätzt, dass das Marfan-Syndrom 1 von 3.000 bis 5.000 Geburten betrifft und unterschiedslos zwischen Männern und Frauen auftritt. Statistiken zeigen, dass 75% der Patienten eine positive Familienanamnese haben; bei den restlichen 25 % liegt die Ursache in sporadischen Mutationen, die in irgendeiner Weise mit dem fortgeschrittenen Alter des Vaters zum Zeitpunkt der Empfängnis in Zusammenhang zu stehen scheinen.
Kinder mit extrem schweren Formen des Marfan-Syndroms haben eine "Lebenserwartung von weniger als einem Jahr".
Vor der Entwicklung chirurgischer Strategien am offenen Herzen hatten die meisten Patienten mit Marfan-Syndrom eine durchschnittliche Lebenserwartung von 32 Jahren; Dank der ständigen Verbesserung medizinischer und pharmakologischer Therapien leben derzeit Betroffene des Marfan-Syndroms im Durchschnitt bis zu 60 Jahre.
Anzeichen und Symptome
Weitere Informationen: Marfan-Syndrom-Symptome
Das Marfan-Syndrom kann völlig asymptomatisch auftreten. Betroffene Patienten haben eine übertrieben schlanke Struktur, sind überproportional groß und dünn. Die unteren und oberen Gliedmaßen sind viel länger als der Rumpf (Dolikostenomegalie). Es ist auch die Rede von Arachnodaktylie um das Konzept der übertriebenen Fingerlänge, typisch für die vom Marfan-Syndrom Betroffenen, am besten auszudrücken: Die Hände werden daher mit den Beinen einer Spinne verglichen.
In Bezug auf die Körpergröße haben diese Patienten eine Statur mit einem Durchschnitt über dem 97. Perzentil.
Unter den anderen charakteristischen Merkmalen, die häufig bei Patienten mit Marfan-Syndrom auftreten, erinnern wir uns auch an:
- Öffnung der Arme größer als die Höhe
- Lockere Gelenke → überhöhte Gelenkbeweglichkeit
- Brustwanddeformität
- Verschiebung des Objektivs
- Oberkörper weniger entwickelt als der untere Bereich
- Spontaner Pneumothorax (11%)
- Skoliose
- Hautstreifen in Höhe von Oberschenkel, Rücken, Deltamuskel, Brust
Unter den problematischsten Anzeichen im Zusammenhang mit dem Marfan-Syndrom erinnern wir uns an den Vorfall der Herzklappe und die Insuffizienz der Mitralklappe: Ein ähnlicher Zustand kann leicht die Erweiterung des Aortenrings und die Aortendissektion begünstigen.
Die Tabelle zeigt die Anzeichen, die bei Patienten mit Marfan-Syndrom gefunden werden können. Die dort beschriebenen Charaktere sind nicht immer vorhanden, aber ein guter Teil davon lässt sich finden.
Mögliche Symptome
Haut
Striae im Brust-, Lenden- und Kreuzbeinbereich
Augen
Sehstörungen, Astigmatismus, Netzhautablösung, Engwinkelglaukom, Linsenluxation, Myopie
Knochenstruktur
Arthralgie, Kyphoskoliose, Dolichostenomelie (übermäßige Entwicklung der Gliedmaßen relativ zum Rumpf), Hypermobilität, hoher Gaumen, deformierte Brust, Plattfüße, enge und dünne Handgelenke, abnormaler Wiedereintritt / Vorwölbung des Brustbeins, Skoliose, gekrümmte Schultern, Spondylolisthesis
Finger
Arachnodaktylie
Lunge
Spontaner Pneumothorax, Dyspnoe, idiopathische obstruktive Lungenerkrankung
Gesichtsveränderungen
Ogivaler Gaumen (Fehlbildung des Gaumens), mandibuläre Retrognathie (Entwicklungsfehler des Kiefers), verlängertes Gesicht
Herz
Angina pectoris, Bauchaortenaneurysma, Herzrhythmusstörungen, Brustaortenerweiterung / -ruptur / -dissektion, Aorteninsuffizienz, Mitralklappenprolaps
Sprache
Schwierigkeiten beim Sprechen
Diagnose
Angesichts der über 200 möglichen Mutationen ist der Einsatz genetischer Marker für diagnostische Zwecke nahezu unmöglich.
Die Beurteilung des Marfan-Syndroms ist nicht immer so unmittelbar, da die phänotypische Ausprägung der Mutation nicht immer offensichtlich und leicht zu identifizieren ist. Die Verzögerung der Diagnose kann das Überleben des Patienten ernsthaft gefährden: Denken Sie zum Beispiel an das Nichterkennen eines Herz-Kreislauf-Problems.
Die diagnostischen Kriterien für das Marfan-Syndrom wurden 1996 international aufgestellt: Die Diagnose besteht in der „Erhebung der Familienanamnese verbunden mit einer Kombination von Haupt- und Nebenindikatoren des Syndroms.
Einige der zahlreichen diagnostischen Tests, die verwendet werden, sind:
- Echokardiogramm
- magnetische Angioresonanz und CT (zur Untersuchung der Aorta)
- Magnetresonanzangiographie (MRA) mit Kontrastmittel (um die inneren Strukturen der Aorta hervorzuheben)
- Untersuchung mit Spaltlampen (zur Analyse einer möglichen Linsenversetzung)
- Augendruckmessung (um das mögliche Vorhandensein eines Glaukoms hervorzuheben)
- Gentests (empfohlen vor der Empfängnis eines Kindes, um festzustellen, ob das Syndrom vorliegt oder nicht)
Therapien
Da es sich um eine genetisch bedingte Krankheit handelt, gibt es kein Medikament oder eine Behandlung, die die Krankheit rückgängig machen kann.
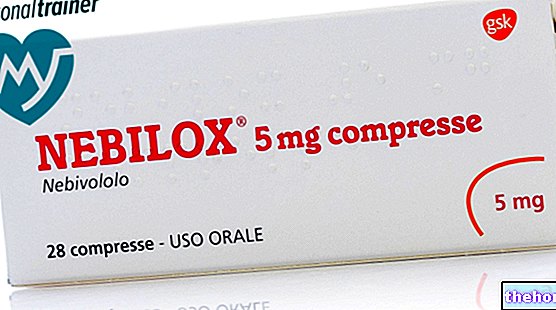
Zur Linderung der Beschwerden und zur Vermeidung von Komplikationen, insbesondere kardialen Komplikationen, ist jedoch der Einsatz von Medikamenten unabdingbar.Hierfür eignen sich besonders blutdrucksenkende Medikamente wie Sartane (vor allem), ACE-Hemmer und Betablocker.
Im Rahmen des Marfan-Syndroms können auch Patienten, die an einer Skoliose leiden, sowie für Glaukombetroffene gezielt behandelt werden.
Eine Operation ist denkbar, um die abnorme Aortenerweiterung zu korrigieren, ein Element, das oft die Mehrheit der Patienten mit Marfan-Syndrom vereint.
Weiter: Marfan-Syndrom - Medikamente und Behandlung "