Allgemeinheit
Als Amyloidose bezeichnet man eine Gruppe von Erkrankungen, die durch die Akkumulation, oft im extrazellulären Bereich, eines als Amyloid bezeichneten fibrillären Proteinmaterials gekennzeichnet sind.Unlösliche Amyloidfibrillen bilden in zahlreichen Organen besonders stabile Ablagerungen.

Andere Fotos Amyloidose - Galerie 2
Symptome und Schwere der Erkrankung hängen von dem überwiegend von der Amyloidakkumulation betroffenen Organ und der Art der Amyloidose ab. Die meisten Fälle sind jedoch systemisch. Das heißt, fibrilläre Ablagerungen sind weit verbreitet und können potenziell die Funktion vieler Gewebe und Organe des Körpers beeinträchtigen . Die Diagnose wird durch eine Biopsie definiert, indem eine kleine Gewebeprobe unter einem Mikroskop untersucht wird. Potenzielle ätiologische Faktoren variieren je nach Amyloidose-Variante. Verfügbare Behandlungen können helfen, die Symptome zu behandeln und die Amyloidproduktion zu begrenzen.
Eigenschaften von Amyloidablagerungen
Amyloidose entsteht durch Störungen der Sekundärstruktur von Proteinen (mit β-Faltblatt-Konfiguration).Unter normalen Bedingungen werden Proteine € tatsächlich in einer linearen Kette von Aminosäuren synthetisiert, die gefaltet eine spezifische räumliche Konformation einnimmt (Proteinfaltung). Dank seiner Struktur, also durch die korrekte Proteinfaltung, kann das Protein die physiologischen Funktionen erfüllen, für die es verantwortlich ist. Die Amyloidproteine stammen von einem Vorläufer ab, der von den Zellen falsch verarbeitet wird (durch "Fehlfaltung" "). Die Proteine, die Fibrillen bilden, sind nach Größe, Aminosäuresequenz und nativer Struktur diversifiziert, werden jedoch zu unlöslichen Aggregaten, die in Struktur und Eigenschaften ähnlich sind. Die Vorläufer von Fibrillen werden durch Primärmoleküle repräsentiert (Beispiel: leichte Kette von Immunglobulinen, β2-Mikroglobulin, Apolipoprotein A1, etc.) oder aus Produkten, die eine Veränderung des s . widerspiegeln Aminosäuresequenz. Die abweichende Sekundärstruktur prädisponiert zur Bildung von Fibrillen, die sich lokal in Geweben und Organen ablagern und zu einer Beeinträchtigung ihrer normalen physiologischen Funktion führen.Mehr als 20 verschiedene Proteinvorstufen wurden identifiziert, die eine amyloide Konformation annehmen können, die warum es viele verschiedene Arten von Amyloidose gibt.
Basierend auf der Lage der Amyloidablagerungen kann die Krankheit unterteilt werden in:
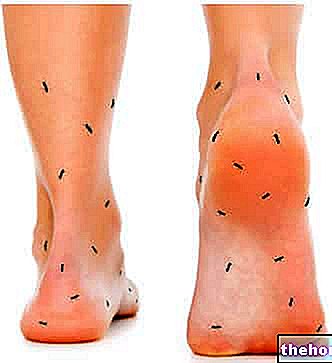
- Lokalisierte Form: Beschränkt auf ein bestimmtes Organ oder Gewebe (Herz, Nieren, Magen-Darm-Trakt, Nervensystem und Dermis) und ist normalerweise weniger schwerwiegend als systemische (diffuse) Formen. Amyloidose kann beispielsweise nur die Haut betreffen und zu Verfärbungen und / oder Juckreiz führen.Eine bestimmte Art von Amyloid-Protein wurde auch im Gehirn von Patienten mit Alzheimer-Krankheit gefunden. Eine lokalisierte Amyloidose ist typisch für Seneszenz und betroffene Patienten. ab Typ-2-Diabetes (wo sich Protein in der Bauchspeicheldrüse ansammelt).
- Systemische Form: Amyloidablagerungen sind in verschiedenen Organen vorhanden und erkennen im Allgemeinen neoplastischen, entzündlichen, genetischen oder iatrogenen Ursprung. Die systemische Amyloidose ist oft sehr schwerwiegend: Sie schädigt häufig Herz, Nieren, Darm und Nerven und verursacht eine fortschreitende Insuffizienz des Organs.
Einstufung
Es gibt viele Formen der Amyloidose, die nach der Natur der Proteine, aus denen die fibrillären Ablagerungen bestehen, klassifiziert werden.
Die gängigsten Varianten sind:
- Primäre Amyloidose (auch Leichtketten-Amyloidose, AL genannt);
- Sekundäre Amyloidose (auch erworbene Amyloidose, AA genannt);
- erbliche Amyloidose;
- Altersbedingte Amyloidose (oder senile systemische Amyloidose).




















.jpg)