Allgemeinheit
Mitochondriale DNA oder mtDNA ist die Desoxyribonukleinsäure, die sich in den Mitochondrien befindet, dh den Organellen eukaryotischer Zellen, die für den sehr wichtigen zellulären Prozess der oxidativen Phosphorylierung verantwortlich sind.

Es weist jedoch auch einige strukturelle und funktionelle Besonderheiten auf, die es in seiner Art einzigartig machen. Zu diesen Besonderheiten gehören: die Zirkularität des Nukleotiddoppelstrangs, der Gengehalt (der nur 37 Elemente beträgt) und das fast vollständige Fehlen nicht kodierender Nukleotidsequenzen.
Mitochondriale DNA erfüllt eine grundlegende Funktion für das Überleben von Zellen: Sie produziert die Enzyme, die für die Durchführung der oxidativen Phosphorylierung notwendig sind.
Was ist mitochondriale DNA?
Mitochondriale DNA oder mtDNA ist die DNA, die sich in den Mitochondrien befindet.
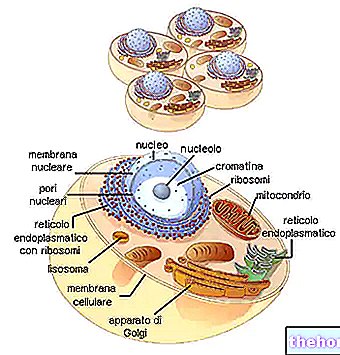
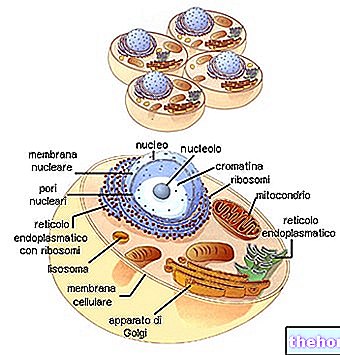
Mitochondrien sind die für eukaryontische Organismen typischen großen Zellorganellen, die die in der Nahrung enthaltene chemische Energie in ATP umwandeln, eine Energieform, die von Zellen verwertet werden kann.
HINTERGRUND ZUR STRUKTUR UND FUNKTION VON MITOCHONDRON
Röhrenförmige, filamentöse oder körnige Mitochondrien befinden sich im Zytoplasma und nehmen fast 25% des Volumens des letzteren ein.
Sie haben zwei Phospholipid-Doppelschichtmembranen, eine weitere externe und eine weitere interne.
Die äußerste Membran, die als äußere mitochondriale Membran bekannt ist, stellt den Umfang jedes Mitochondriums dar und verfügt über Transportproteine (Porine und mehr), die sie für Moleküle mit einer Größe von 5.000 Dalton oder weniger durchlässig machen.
Die innerste Membran, die als innere mitochondriale Membran bekannt ist, enthält alle enzymatischen (oder enzymatischen) und Coenzym-Komponenten, die für die Synthese von ATP notwendig sind, und definiert einen zentralen Raum, die sogenannte Matrix.
Im Gegensatz zur äußersten Membran weist die innere Mitochondrienmembran zahlreiche Einstülpungen – die sogenannten Ridges – auf, die ihre Gesamtfläche vergrößern.
Zwischen den beiden mitochondrialen Membranen befindet sich ein Raum von fast 60-80 Angström (A).Dieser Raum wird als Intermembranraum bezeichnet. Der Intermembranraum hat eine Zusammensetzung, die der des Zytoplasmas sehr ähnlich ist.
Die von den Mitochondrien betriebene Synthese von ATP ist ein sehr komplexer Vorgang, den Biologen mit dem Begriff oxidative Phosphorylierung identifizieren.
PRÄZISE LAGE DER MITOCHONDRALEN DNA UND MENGE

Abbildung: Ein menschliches Mitochondrium.
Mitochondriale DNA befindet sich in der mitochondrialen Matrix, d. h. in dem Raum, der von der inneren Mitochondrienmembran begrenzt wird.
Nach zuverlässigen wissenschaftlichen Studien kann jedes Mitochondrium 2 bis 12 Kopien mitochondrialer DNA enthalten.
Angesichts der Tatsache, dass einige Zellen im menschlichen Körper mehrere Tausend Mitochondrien enthalten können, kann die Gesamtzahl der Kopien der mitochondrialen DNA in einer einzelnen menschlichen Zelle bis zu 20.000 Einheiten erreichen.
bitte beachten Sie: Die Anzahl der Mitochondrien in menschlichen Zellen variiert je nach Zelltyp. Zum Beispiel können Hepatozyten (d. h. Leberzellen) jeweils zwischen 1.000 und 2.000 Mitochondrien enthalten, während Erythrozyten (d. h. rote Blutkörperchen) völlig frei davon sind.
Struktur
Die allgemeine Struktur eines mitochondrialen DNA-Moleküls ähnelt der allgemeinen Struktur der nuklearen DNA, dh dem genetischen Erbe, das im Kern eukaryontischer Zellen vorhanden ist.
In der Tat, analog zur Kern-DNA:
- Mitochondriale DNA ist ein Biopolymer, das aus zwei langen Nukleotidsträngen besteht. Nukleotide sind organische Moleküle, die aus der Vereinigung von drei Elementen resultieren: einem Zucker mit 5 Kohlenstoffatomen (im Fall von DNA Desoxyribose), einer stickstoffhaltigen Base und einer Phosphatgruppe.
- Jedes Nukleotid der mitochondrialen DNA bindet über eine Phosphodiesterbindung zwischen dem Kohlenstoff 3 seiner Desoxyribose und der Phosphatgruppe des unmittelbar folgenden Nukleotids an das nächste Nukleotid desselben Strangs.
- Die beiden Stränge der mitochondrialen DNA haben entgegengesetzte Orientierungen, wobei das Ende des einen mit dem Ende des anderen wechselwirkt und umgekehrt.Diese besondere Anordnung wird als antiparallele Anordnung (oder antiparallele Orientierung) bezeichnet.
- Die beiden Stränge der mitochondrialen DNA interagieren miteinander durch die stickstoffhaltigen Basen.
Insbesondere bildet jede stickstoffhaltige Base jedes Filaments Wasserstoffbrückenbindungen mit einer und nur einer stickstoffhaltigen Base, die auf dem anderen Filament vorhanden ist.
Diese Art der Wechselwirkung wird "Paarung zwischen stickstoffhaltigen Basen" oder "Paar von stickstoffhaltigen Basen" genannt. - Die stickstoffhaltigen Basen der mitochondrialen DNA sind Adenin, Thymin, Cytosin und Guanin.
Die Paarung, zu der diese stickstoffhaltigen Basen führen, ist nicht zufällig, sondern hochspezifisch: Adenin interagiert nur mit Thymin, während Cytosin nur mit Guanin interagiert. - Mitochondriale DNA beherbergt Gene (oder Gensequenzen). Gene sind Sequenzen von mehr oder weniger langen Nukleotiden mit einer genau definierten biologischen Bedeutung. In den meisten Fällen bilden sie Proteine.

STRUKTURELLE BESONDERHEITEN DER MITOCHONDRALEN DNA
Abgesehen von den oben genannten Analogien weist die menschliche mitochondriale DNA einige strukturelle Besonderheiten auf, die sie erheblich von der menschlichen nuklearen DNA unterscheidet.
Erstens ist es ein zirkuläres Molekül, während die nukleäre DNA ein lineares Molekül ist.
Somit hat es 16.569 stickstoffhaltige Basenpaare, während die Kern-DNA satte 3,3 Milliarden hat.
Es enthält 37 Gene, während die Kern-DNA zwischen 20.000 und 25.000 zu enthalten scheint.
Es ist nicht in Chromosomen organisiert, während die Kern-DNA in 23 Chromosomen unterteilt ist und mit einigen spezifischen Proteinen eine Substanz namens Chromatin bildet.
Schließlich umfasst es eine Reihe von Nukleotiden, die gleichzeitig an zwei Genen beteiligt sind, während die nukleäre DNA Gene hat, deren Nukleotidsequenzen gut definiert und voneinander verschieden sind.
Herkunft
Mitochondriale DNA hat höchstwahrscheinlich einen "bakteriellen" Ursprung.
Tatsächlich glauben Molekularbiologen auf der Grundlage zahlreicher unabhängiger Studien, dass das zelluläre Vorhandensein von mitochondrialer DNA das Ergebnis des Einbaus unabhängiger bakterieller Organismen, die den Mitochondrien sehr ähnlich sind, durch angestammte eukaryontische Zellen ist.
Diese merkwürdige Entdeckung hat die wissenschaftliche Gemeinschaft nur teilweise überrascht, da die in Bakterien vorhandene DNA im Allgemeinen ein zirkulärer Nukleotidstrang ist, wie die mitochondriale DNA.
Die Theorie, nach der Mitochondrien und mitochondriale DNA einen „bakteriellen Ursprung“ haben, hat den Namen „endosymbiotische Theorie“, von dem Wort „Endosymbiose“. „Einverleibung des einen in das andere, um einen gewissen Vorteil zu erlangen.
Neugier
Nach zuverlässigen wissenschaftlichen Studien hätten im Laufe der Evolution viele bakterielle Gene, die auf der zukünftigen mitochondrialen DNA vorhanden sind, ihren Standort gewechselt und sich in die nukleäre DNA bewegt.
Mit anderen Worten, zu Beginn der Endosymbiose befanden sich einige Gene, die jetzt auf der nuklearen DNA vorhanden sind, in der DNA dieser bakteriellen Organismen, die später zu Mitochondrien werden sollten.
Um die Theorie bezüglich einer Verschiebung von Genen zwischen mitochondrialer DNA und nuklearer DNA zu untermauern, ist die Beobachtung, dass bestimmte Gene bei einigen Arten von mitochondrialer DNA und bei anderen von nuklearer DNA abstammen.
Funktion
Mitochondriale DNA produziert Enzyme (dh Proteine), die für die korrekte Durchführung des heiklen oxidativen Phosphorylierungsprozesses notwendig sind.
Die Anweisungen zur Synthese dieser Enzyme liegen in den 37 Genen, aus denen das mitochondriale DNA-Genom besteht.
WELCHER MITOCHONDRALER DNA-GENCODE: DIE DETAILS
Die 37 Gene der mitochondrialen DNA kodieren für: Proteine, tRNA und rRNA.
Bestimmtes:
- 13 kodieren für 13 Proteine, die für die Durchführung der oxidativen Phosphorylierung verantwortlich sind
- 22 Code für 22 tRNA-Moleküle
- 2 kodieren 2 rRNA-Moleküle
Die Moleküle der tRNA und rRNA sind von grundlegender Bedeutung für die Synthese der oben genannten 13 Proteine, da sie die Maschinerie bilden, die ihre Produktion reguliert.
Mit anderen Worten, die mitochondriale DNA besitzt die Informationen, um einen bestimmten Satz von Proteinen und die für ihre Synthese notwendigen Werkzeuge zu produzieren.
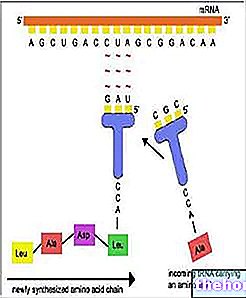
Was sind RNA, tRNA und rRNA?
RNA oder Ribonukleinsäure ist die Nukleinsäure, die ausgehend von der DNA eine grundlegende Rolle bei der Bildung von Proteinen spielt.
Im Allgemeinen einzelsträngig, kann das KNN in verschiedenen Formen (oder Typen) existieren, abhängig von der spezifischen Funktion, an die es delegiert wird.
TRNA und rRNA sind zwei dieser möglichen Formen.
Die tRNA wird verwendet, um bei der Herstellung von Proteinen Aminosäuren hinzuzufügen.Aminosäuren sind die molekularen Einheiten, aus denen Proteine bestehen.
Die rRNA bildet die Ribosomen, also die zellulären Strukturen, in denen die Proteinsynthese stattfindet.
Um das KNN und seine Funktionen im Detail kennenzulernen, können die Leser hier klicken.
FUNKTIONELLE DETAILS DER MITOCHONDRALEN DNA
Aus funktioneller Sicht weist die mitochondriale DNA einige besondere Merkmale auf, die sie deutlich von der nuklearen DNA unterscheiden.
Hier ist, woraus diese besonderen Eigenschaften bestehen:
- Mitochondriale DNA ist insofern halbunabhängig, als sie die Intervention einiger Proteine benötigt, die aus nuklearer DNA synthetisiert werden.
Andererseits ist die nukleare DNA völlig autonom und produziert selbst alles, was sie zur ordnungsgemäßen Erfüllung ihrer Aufgaben benötigt. - Mitochondriale DNA hat einen etwas anderen genetischen Code als nukleäre DNA. Dies führt zu einer Reihe von Unterschieden bei der Herstellung von Proteinen: Wenn eine bestimmte Nukleotidsequenz in der nuklearen DNA zur Bildung eines bestimmten Proteins führt, führt die gleiche Sequenz in der mitochondrialen DNA zur Bildung eines etwas anderen Proteins.
- Mitochondriale DNA hat sehr wenige nicht-kodierende Nukleotidsequenzen, dh sie produziert keine Proteine, tRNAs oder rRNAs. Prozentual ausgedrückt sind nur 3% der mitochondrialen DNA nicht kodierend.
Andererseits ist die nukleäre DNA nur zu 7% kodierend, enthält also viele nicht kodierende Nukleotidsequenzen (bis zu 93%).
Tabelle: Zusammenfassung der Unterschiede zwischen menschlicher mitochondrialer DNA und menschlicher nuklearer DNA.
Mitochondriale DNA
Kern-DNA
- Es ist kreisförmig
- Es ist linear
- Es hat insgesamt 16.569 stickstoffhaltige Basenpaare
- Es hat insgesamt 3,3 Milliarden stickstoffhaltige Basenpaare
- Es enthält insgesamt 37 Gene
- Es enthält zwischen 20.000 und 25.000 Gene
- Um richtig zu funktionieren, braucht es die Unterstützung einiger Genprodukte, die aus der nuklearen DNA stammen
- Es ist autonom und produziert selbst alles, was es braucht, um seine Funktionen richtig zu erfüllen
- Es kann in jedem einzelnen Mitochondrium in mehreren Kopien vorhanden sein
- Es ist einzigartig, das heißt, es ist nur in einer Kopie vorhanden und befindet sich im Kern
- 97% der Nukleotidsequenz, aus der es besteht, kodiert
- Nur 7% der Nukleotidsequenz, aus der es besteht, kodiert
- Es ist nicht in Chromosomen organisiert
- Es ist in 23 Chromosomen unterteilt
- Es verwendet einen genetischen Code, der sich etwas vom "traditionellen" Code unterscheidet
- Verwenden Sie den "traditionellen" genetischen Code
- Sein Erbe ist mütterlicherseits
- Sein Erbe ist halb mütterlich und halb väterlich
- Einige seiner Nukleotide sind gleichzeitig an zwei Genen beteiligt
- Die Nukleotidsequenzen, aus denen die Gene bestehen, sind gut voneinander zu unterscheiden
Nachlass
Die mitochondriale DNA-Vererbung ist streng mütterlicherseits.
Das bedeutet, dass bei einem Elternpaar die Frau die mitochondriale DNA an die Nachkommen (also an die Kinder) weitergibt.
Im Gegensatz dazu ist die Vererbung der nuklearen DNA halb mütterlich und halb väterlich, dh beide Eltern tragen gleichermaßen zur Übertragung der nuklearen DNA bei den Nachkommen bei.
bitte beachten Sie: Die mütterliche Vererbung der mitochondrialen DNA umfasst auch die mitochondriale Struktur. Daher sind die in einem Individuum vorhandenen Mitochondrien mütterlicherseits.
Assoziierte Pathologien
Prämisse: Eine genetische Mutation ist eine dauerhafte Veränderung der Sequenz von Nukleotiden, aus denen ein nukleäres oder mitochondriales DNA-Gen besteht.
Typischerweise führt das Vorhandensein einer genetischen Mutation zu einer "Veränderung oder einem Verlust der normalen Funktion des betroffenen Gens.
Das Vorhandensein von Mutationen in den mitochondrialen DNA-Genen kann zu einer Vielzahl von Krankheiten führen, darunter:
- Lebersche hereditäre Optikusneuropathie
- Kearns-Sayre-Syndrom
- Leigh-Syndrom
- Der Mangel an Cytochrom-C-Oxidase
- Progressive externe Ophthalmoplegie
- Pearson-Syndrom
- Mitochondriale Enzephalomyopathie mit Laktatazidose und Schlaganfall-ähnlichen Episoden (MELAS-Syndrom)
- Diabetes mit durch die Mutter übertragener Taubheit
- Myoklonische Epilepsie mit unregelmäßigen roten Fasern
Hinsichtlich der pathologischen Zustände, die mit einer oder mehreren mitochondrialen DNA-Mutationen verbunden sind, müssen zwei Aspekte geklärt werden.
Erstens hängt die Schwere der Erkrankung von der quantitativen Beziehung zwischen mutierten mitochondrialen DNAs und gesunden, normalen mitochondrialen DNAs ab. Wenn die Anzahl der mutierten mitochondrialen DNAs erheblich größer ist als die der gesunden DNAs, wird der resultierende Zustand schwerwiegender sein.
Zweitens betreffen Mutationen in der mitochondrialen DNA nur einige Gewebe des Organismus, insbesondere solche, die aufgrund des oxidativen Phosphorylierungsprozesses große Mengen an ATP benötigen.Das ist durchaus verständlich: Mehr als eine Fehlfunktion der mitochondrialen DNA zu erleiden, sind die Zellen, die am meisten benötigt werden die Funktion, die die mitochondriale DNA normalerweise erfüllt.
LEBERS ERBLICHE OPTISCHE NEUROPATHIE
Lebers hereditäre Optikusneuropathie entsteht durch die Mutation von bis zu vier mitochondrialen DNA-Genen. Diese Gene enthalten die Informationen, die zur Synthese des sogenannten Komplexes I (oder NADH-Oxid-Reduktase) führen, eines der verschiedenen Enzyme, die am oxidativen Phosphorylierungsprozess beteiligt sind.
Die Manifestationen der Pathologie bestehen in einer fortschreitenden Degeneration des Sehnervs und einem allmählichen Verlust des Sehvermögens.
KEARNS-SAYRE-SYNDROM
Das Kearns-Sayre-Syndrom tritt aufgrund des Fehlens einer angemessenen Menge an mitochondrialer DNA auf (Hinweis: das Fehlen einer bestimmten Nukleotidsequenz wird als Deletion bezeichnet).
Menschen mit Kearns-Sayre-Syndrom entwickeln Ophthalmoplegie (totale oder teilweise Lähmung der Augenmuskeln), eine Form von Retinopathie und Herzrhythmusstörungen (atrioventrikulärer Block).
LEIGH-SYNDROM
Das Leigh-Syndrom entsteht durch mitochondriale DNA-Mutationen, die das ATP-Synthase-Protein (auch V-Komplex genannt) und/oder einige tRNAs beeinflussen können.
Das Leigh-Syndrom ist eine fortschreitende neurologische Erkrankung, die im Säuglings- oder Kindesalter auftritt und verantwortlich ist für: Entwicklungsverzögerung, Muskelschwäche, periphere Neuropathie, motorische Störungen, Atembeschwerden und Ophthalmoplegie.
MANGEL AN CYTOCHROM-COXIDASE
Ein Cytochrom-C-Oxidase-Mangel tritt aufgrund der Mutation von mindestens 3 mitochondrialen DNA-Genen auf. Diese Gene sind für die korrekte Synthese des Cytochrom-C-Oxidase-(oder Komplex-IV-)Enzyms essentiell, das am oxidativen Phosphorylierungsprozess beteiligt ist.
Typische Manifestationen eines Cytochrom-C-Oxidase-Mangels sind: Skelettmuskelfunktionsstörung, Herzfunktionsstörung, Nierenfunktionsstörung und Leberfunktionsstörung.
PROGRESSIVE EXTERNE OPHTHALMOPLEGIE
Progressive externe Ophthalmoplegie entsteht durch das Fehlen einer erheblichen Anzahl von mitochondrialen DNA-Nukleotiden (Deletion)
Mit progressivem Charakter (wie der Name vermuten lässt) verursacht diese Pathologie eine Lähmung der Augenmuskeln mit nachfolgender Ptosis und erheblichen Sehproblemen.
PEARSON-SYNDROM
Das Pearson-Syndrom tritt nach einer auffälligen Deletion mitochondrialer DNA auf, ähnlich der progressiven externen Ophthalmoplegie und dem Kearns-Sayre-Syndrom.
Typische Manifestationen des Pearson-Syndroms sind: sideroblastische Anämie, Pankreasdysfunktion (zB insulinabhängiger Diabetes), neurologische Ausfälle und Muskelerkrankungen.
Das Pearson-Syndrom führt in der Regel dazu, dass die betroffene Person in jungen Jahren stirbt. Tatsächlich erreichen diejenigen, die von dieser Pathologie betroffen sind, selten das Erwachsenenalter.
MELAS-SYNDROM
Das MELAS-Syndrom, auch bekannt als mitochondriale Enzephalomyopathie mit Laktatazidose und Schlaganfall-ähnlichen Episoden, entsteht durch die Mutation von mindestens 5 mitochondrialen DNA-Genen.
Diese Gene tragen zur Synthese von NADH-Oxid-Reduktase oder Komplex I und einiger tRNAs bei.
Das MELAS-Syndrom beinhaltet das Vorhandensein von neurologischen Störungen, Muskelstörungen, ungewöhnlicher Ansammlung von Milchsäure im Gewebe (mit allen Begleitsymptomen), Atemproblemen, Verlust der Kontrolle der Darmfunktion, wiederkehrender Müdigkeit, Nierenproblemen, Herzproblemen, Diabetes , Epilepsie und Mangel an Koordination.
ANDERE PATHOLOGIEN
Verschiedenen wissenschaftlichen Studien zufolge würden auch Krankheiten wie das zyklische Erbrechen, Retinitis pigmentosa, Ataxie, Parkinson-Krankheit und Alzheimer-Krankheit eine Beteiligung der mitochondrialen DNA und einiger ihrer Mutationen aufweisen.